
Duchenne Muscular Dystrophy
Introduction
 |
| Image courtesy of Symbiosis Online Publishing Opens in new window
Duchenne muscular dystrophy (DMD), named after a French neurologist Guillaume Duchenne, who published a series of illustrated articles in 1868 that described key features of the disease, is the most common form of muscular dystrophy Opens in new window in children, characterized by muscle wasting, weakness, and progressive loss of contractile function. |
Duchenne muscular dystrophy DMD is primarily a skeletal and heart muscle disease that makes walking and breathing difficult, it can also affect cognition Opens in new window.
DMD is inherited as an X-linked recessive trait Opens in new window, affecting approximately 1 in 3500 live-born male infants; making it one of the more common genetic diseases among all populations.
Classic DMD usually presents clinically in the first 5 years of life, with delayed motor skills Opens in new window or an abnormal gait, followed by progressive muscle weakness.
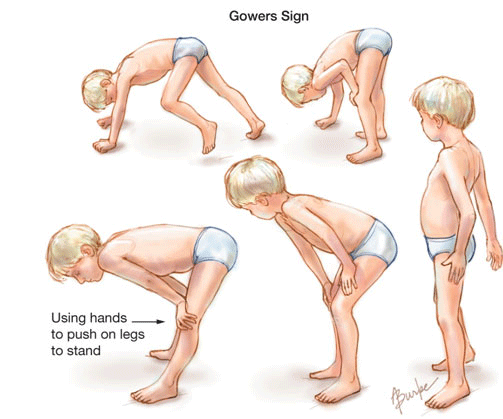
Proximal muscles are involved initially, resulting in a characteristic waddling gait and Gowers’s sign (in which patient use their hands and arms to raise the upper body when rising from a lying position [see image above]).
Distal muscles become progressively involved, and muscles (particularly the calves) may exhibit pseudohypertrophy because of increased fat and connective tissue. Serum creatine kinase levels are elevated until muscle mass becomes decreased.
Progressive weakness initially affects the lower extremities, causing most DMD patients to require wheelchairs between 10 and 15 years of age. Continual degeneration and regeneration and inflammation of muscle eventually lead to the replacement of muscle tissue by adipose and connective tissue.
Scoliosis is common and affects respiratory function. Chronic respiratory insufficiency develops in all patients. Cardiorespiratory failure is the primary cause of death.
DMD is caused by the deficiency of a protein component of muscle tissue called dystrophin.
Dystrophin is part of the membrane cytoskeleton in normal muscle, and a deficiency leads to myofiber necrosis, presumably because of membrane instability.
Whereas 70% of cases are inherited through a carrier mother, approximately 30% of cases occur without previous family history, representing apparent de novo mutations in the dystrophin gene.
Clinical management of patients with DMD is complex, requiring a multidisciplinary team approach. In addition to healthcare professionals, team members in the areas of psychosocial, gastrointestinal, pain, and speech and language are required.
Because DMD is an X-linked recessive disorder Opens in new window, most carrier females are asymptomatic. Similar to other X-linked diseases, the varying degree of clinical manifestations among carrier females depends on the degree of inactivation of the X chromosome Opens in new window harboring the mutant DMD gen in various tissues where the DMD protein is expressed.
Up to as many as 20% of carriers can display some symptoms of DMD. Most frequently observed is muscle weakness with elevated serum CK levels or cardiac involvement.
Genetic and Biochemical Characteristics
The genetic defects associated with DMD include deletions or non-sense mutations Opens in new window in the dystrophin gene.
The skeletal muscle isoform of dystrophin Opens in new window is a rodlike cytoskeletal protein localized to the inner surface of the skeletal muscle plasma membrane, the inner layer of the sarcolemma (Sanes, 2004). It forms part of the dystrophin-glycoprotein complex Opens in new window (DGC) linking the cytoskeleton and the extracellular matrix.
The DGC is also called the dystrophin-associated protein complex (DAP), as not all proteins in the complex are glycosylated (Ozawa, 2004).
The specific function(s) of dystrophin and the DGC are not completely known, although it is believed that they could serve both mechanical and signaling roles (Durbeej and Campbell, 2002; Roberts, 2001).
The absence of dystrophin Opens in new window could render the membrane susceptible to mechanical injury, leaky to Ca2+, or susceptible to disrupted signaling. These effects alone or in combination could dramatically disrupt muscle function.
Dystrophin-deficient muscle is characterized by increased permeability to endogenenous macromolecules flowing out of and into the muscle cell.
A classical marker for DMD is an elevated serum muscle creatine kinase concentration. In addition, muscle fibers stain positively for endogenous extracellular proteins such as albumin and immunoglobulins such as IgG and IgM.
These clinical features suggest that the membranes of dystrophic muscle cells are fragile and leaky, and that this contributes to the progressive force loss and ultimate death of the muscle cell.
In DMD, the protein dystrophin and the associated proteins of the DGC are absent, and this leads to severe pathophysiology of skeletal and cardiac muscle.
The two main features of DMD pathophysiology are:
- progressive degeneration of muscle tissue with replacement by noncontractile fat and connective tissues and
- progressive muscle weakness.
The disease process is characterized by progressive rounds of myofiber necrosis followed by rounds of muscle fiber regeneration. Fiber regeneration is identified histologically based on the presence of centralized nuclei.
Grouped degenerating and necrotic fibers are characteristic of DMD muscle biopsies even before muscle weakness is clinically observed, with evidence of myofiber degeneration in boys at approximately age 3 years.
Ultimately, the combination of progressive fibrosis and muscle fiber loss results in muscle wasting, weakness, and failure. Much has yet to be learned about both the pathophysiological mechanisms of DMD, and about why repair processes are ineffective.
Clinical Features
Duchenne muscular dystrophy is characterized by childhood onset, a prevalence in boys, occurrence in children within the same family, progressive muscle weakness yet a gradual increase in size of many affected muscles (pseudohypertrophy), and abundant fibrosis and adipose tissue at later stages of the disease.
The onset of DMD is evident at approximately age 3 to 5 years. One-third of DMD patients show intellectual impairment, and both speech and motor skill Opens in new window development are delayed.
For example, walking is delayed approximately five months compared to that in a normal child. The muscle involvement is bilateral and symmetrical and affects the lower limbs first.
A common clinical sign is pseudohypertrophy, in which muscles such as the gastrocnemii, or calf muscles, swell and appear larger than normal.
The proximal muscles are more affected than the distal muscles, with contractures evident at the elbows, knees, and hips.
Progressive muscle degeneration driven by presently undefined mechanisms compromises the child’s ability to support himself.
With disease progression, the child develops thoracolumbar kyphosis (a prominent outward curve) and scoliosis (a lateral curve) of the spine (Karol, 2007).
Additional features are a waddling gait due to weakness in hip abductors, lumbar lordosis due to weak gluteal muscles, and the need for Gower’s maneuver due to weakened knee and hip extensors.
Gower’s maneuver (as shown in the introductory image) involves using the hands to support the body’s rise to a standing position from the floor or a chair. Eventually the child loses all ability to support himself.
By age 12, 95% of patients need to use a wheelchair. Cardiopulmonary function steadily deteriorates.
By age 20, 90% of DMD patients die, most from cardiac failure and respiratory insufficiency (Emery, 1993).
Treatment
As mentioned earlier, there is presently no effective cure for DMD, and the plan for management of the disease should be individualized.
The goals of treatment for boys with DMD should be consistent with maintaining mobility, maximizing muscle strength performance and pulmonary function, and optimizing performance of activities of daily living.
Treatment plans should be determined by boy’s developmental and functional needs.
Goals of care should be centered on a multidisciplinary approach including specialists from the following areas: clinical genetics, neurology, pulmonology, cardiology, nutrition, rehabilitation medicine, orthopedics, and neuropsychology (American Thoracic Society [ATS], 2004).
Summary of Primary Care Needs for the Child with Duchenne Muscular Dystrophy
Growth and Development
- Close surveillance of growth is necessary, adapting measurement techniques is necessary as disease progresses.
- Monitor growth parameters at every visit with the appropriate measurement and plotting of weight, length or height, head circumference, and BMI.
- Monitor development and need for adaptive equipment
Diet
- Encourage boys to remain as active as possible and to maintain a healthy weight by making healthy food choices.
- Closely monitor weight gain with decreasing mobility and exercise and corticosteroid use.
- Consider a decrease in energy expenditure when calculating daily caloric needs using an appropriate formula: Daily energy intake in kcal = 2000 – Age (in years) x 50, or 9 to 11 kcal/Height (in cm), and individualize plans as necessary.
- Consider calcium and vitamin D supplementation when on corticosteroid therapy.
- Diet modifications and adaptive equipment needed with disease progression.
Safety
- Instruct families in keeping both the home and school environment safe and clear from objects on the floor and sharp edges and corners.
- Assist in arranging a home evaluation to assess for accessibility.
- Suggest adaptations to ensure safety.
- Assist in relocation to accessible housing or an apartment building with an elevator, when necessary.
- Instruct families to notify local police and fire departments to facilitate safety during an emergency.
Immunizations
- Routine immunizations should be given on schedule.
- All family members should be immunized against influenza on an annual basis.
- Administer heptavalent pneumococcal polysaccharide-protein conjugate vaccine (PPCV-7) before age 2, and boys ages 2 and older should receive 23-valent pneumococcal polysaccharide vaccine (PPCV-23) as recommend for age and immunization status.
- Systemic corticosteroid therapy can cause immunosuppression. Empiric guidelines recommend that children receiving low or moderate doses (<2 mg/kg/day of prednisone or its equivalent, or <20 mg/day if weight is >10 kg) can receive live-virus vaccines; however, the AAP guidelines should be referred to if the dose increases, in which cases live vaccines should be avoided.
Screening
- Vision — Routine screening is recommended. Annual pediatric ophthalmologist referral is needed for boys on steroid therapy.
- Mild color-blindness may be present.
- Hearing — Routine screening is recommended.
- Dental —Routine screening and early referral to a dentist is recommended.
- Blood pressure — Screen at every visit after start of corticosteroid treatment.
- Hematocrit — Routine screening for anemia. Annual complete blood count and arterial or free-flowing blood gas for nonambulatory patient is recommended.
- Tuberculosis — Routine screening for boys at increased risk and before the onset of steroid treatment.
See also:
- Aartsma-Rus, A., van Deutekom, J.C.T., Fokkema, I. F., et al. (2006). Entries in the Leiden Duchenne muscular dystrophy database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve, 34 135-144.
- American Academy of Pediatrics (AAP). (2005). Clinical report: Cardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophy: Section cardiology and cardiac surgery. Pediatrics, 116(6), 1569-1573.
- American Thoracic Society (ATS). (2004). American Thoracic Society documents Respiratory care of the patient with Duchenne muscular dystrophy, ATS consensus statement. Am J Respir Crit Care Med, 170, 456-465.
- Biggar, W.D., Harris, V.A., Eliasoph, L., et al. (2006). Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromusc Discord, 16, 249-255.
- Bushby, K., Bourke, J., Bullock, R. et al. (2005). The multidisciplinary management of Duchenne muscular dystrophy. Curr Pediatr, 15, 292-300.
- Foster, K., Foster, H., & Dickson, J. G. (2006). Gene therapy progress and prospects: Duchenne muscular dystrophy. Gene Ther, 13. 1677-1685.
- Kohler, M., Clarenbach, C., Boni, L., et al. (2005). Quality of life, physical disability, and respiratory impairment in Duchenne muscular dystrophy. Am J Respir Crit Care Med, 172(8), 1032-1036.
