
Ehlers–Danlos Syndrome
Clinical Features, Genes and Therapeutics
|
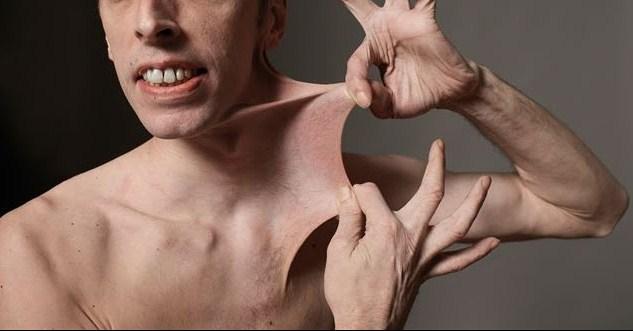
| E–DS' highly elastic skin | Image courtesy of ORTHO BULLETS Opens in new window The Ehlers–Danlos syndrome is a group of collagen disorders of connective tissue, which includes tendons, ligaments, hyperextensible skin, bones, cartilage and the membranes surrounding blood vessels and nerves. These highly variable disorders can be characterized by easily bruised and highly elastic skin that is prone to tear, extreme joint laxity giving the appearance of double–jointedness, and multiple chronic dislocations and broken bones. |
The syndrome is named for eminent Danish dermatologist Eduard Ehlers (1863–1937) and French dermatologist Henri A. Danlos (1844–1912), who described various forms of the condition in the early 1900s.
Ehlers described patients with lax joints and elastic skin in 1901, while Danlos reported unusual scarring and skin fragility in 1908.
Subsequent clinical work has identified at least 10 varieties of Ehlers–Danlos (E–D) syndrome, with most involving the eyes, skin, and joints; and are transmitted in autosomal dominant, autosomal recessive and X-linked forms.
Premature birth is common in some forms, due to early rupture of the fetal membranes. At birth, there are often scars on the forehead and chin. The ears frequently stick outward.
The skin is often described as velvety, or like chamois. Bruised skin heals with peculiar, cigarette paper scars. Normal bumps and bruises can result in serious injury.
In some patients most surgery can be undertaken only at great risk, due to fragile tissue and uncontrollable bleeding, and in some forms of the disorder major heart problems, rupture of major blood vessels, aortic aneurisms and internal bleeding can cause lethal complications.
The forms vary in their severity. Some are associated with mild symptoms. Others can result in severe disability and death, as described above.
Typically, there is little variability among affected family members. In 1997, researchers proposed a simpler classification that reduces the number of major types from the previously delineated 10 down to six.
Cause: Gene Expression
Patients with Ehlers–Danlos syndrome manifests problems that are usually attributed to the production of abnormal collagen, the protein that is the main structural component of the connective tissue.
Because the production of collagen necessitates many biochemical steps that are controlled by several genes, the potential exists for any of these genes to mutate, producing selective defects in collagen synthesis.
Tissue strength and limited elasticity are associated with normal collagen structures. The various forms of abnormal collagen result in many overlapping clinical features for each of the types of the Ehlers–Danlos syndrome.
Types and Gene Description
The remainder of this entry concentrates on the 10 forms of Ehlers–Danlos syndromes, discusses the clinical features and associated genes.
- EDS Type I (gravis)
EDS Type I (gravis) consists in severe classic features as described above of soft, fragile, hyperextensible skin, cigarette paper scars, easy bruisability, and large and small joint hypermobility.
In Type I Ehlers-Danlos syndrome, hernias and varicose veins are frequent. It is an autosomal dominant disorder; at least some cases are caused by mutations in the gene for the alpha-1 chain of a type V collagen (COL5AI).
- EDS Type II (mitis)
Type II (mitis) consists in classic features with milder expression. Scarring, bruising and joint hypermobility are less severe. Varicose veins and hernias are less common. This is the most common variant of EDS. The inheritance pattern is autosomal dominant.
Some cases are allelic (see Allele Opens in new window) to the type I form caused by COL5AI; that is, mitis involves different mutations to the same gene. Types I and II are grouped together in the new classification as classical type.
Less commonly, mutations Opens in new window in the COL5A2, COL1A1 and COL1A2 genes, or even more infrequently in another gene, TNXB, cause the classical type.
- Type III (benign familial hypermobility)
Minimal skin involvement, marked large and small joint hypermobility, recurrent joint dislocations and early osteoarthritis are the hallmarks of this autosomal dominant form.
While in most cases the underlying molecular defect of this very common type of Ehlers-Danlos syndrome is unknown, mutations Opens in new window in one copy of the TNXB gene may be responsible for the condition in 5–10% of cases. This gene encodes a protein called tenascin-X and is located on the short arm of chromosome 6.
- Type IV (ecchymotic or arterial type)
In EDS Type IV, the skin is thin and not hyperextensible, with visible underlying veins prone to easy bruising. Joints are not hypermobile (except small joints of the hand).
Bowel rupture, uterine rupture during pregnancy, arterial rupture (including aneurysms) which may lead to death also occur.
Significant complications are rare before the 20s, but the mean of age death is in the early 30s.
The disorder is autosomal dominant Opens in new window; mutations in the COL3A1 gene result in abnormal type III collagen. New mutations are common: One-half of affected individuals have a family history.
- Type V (X-linked)
The EDS Type V is similar to its type II variant but transmitted as an X-linked recessive Opens in new window trait.
- Type VI (ocular)
This variety of EDS presents with ocular fragility along with skin and joint features similar to type II, e.g., scoliosis (spinal curvature), poor muscle tone and motor development delay.
Its inheritance pattern is autosomal recessive Opens in new window and is caused by a defect in the enzymes lysl oxidase. This form is very rare with fewer than 60 cases having been reported.
- EDS Type VII
The Type VII variant is characterized by soft hyperextensible, easily bruisable skin that is not fragile and with nearly normal scarring. It also presents with marked joint hypermobility and congenital hip dislocation.
EDS Type VII has been subclassified into three types: Types A and B are autosomal dominant and have mutations in the COL1A1 and COL1A2 genes respectively, while type C is autosomal recessive and due to a deficiency of the enzyme procollagen N-proteinase.
- EDS Type VIII (periodontal)
EDS type VIII is a severe generalized periodontal (gum) disease, accompanied with loss of most teeth by the early 20s. This is in addition to its characteristic thin, fragile, hypersensitive skin exhibiting abnormal scarring and easy bruising, and the mild to moderate joint laxity. It is an autosomal dominant disorder.
- EDS Type IX (occipital horns)
The Type IX variant is X-linked Opens in new window, similar to cutis laxa Opens in new window: lax, soft skin, bony growths (exostoses) at the back of the skull (occipital horns) and other skeletal abnormalities. Bladder diverticulae during childhood and mild chronic diarrhea are common.
The X-linked recessive condition results from abnormal copper metabolism. Different mutations in the same gene cause Menkes syndrome. Thus, it is allelic to that disorder.
- EDS Type X
The Type X variant consists in features similar to type II with a disorder of platelet aggregation. Transmitted through autosomal recessive inheritance. A single affected family has been described.
Types I and II together are estimated to affect one in 20,000 to 40,000 people, and type III as many as one in 10,000 to 15,000 people. Type IV occurs in about one in 250,000 people.
The other types are all very rare. Many cases may remain undiagnosed. Physicians may regard the continual bone breakage some affected individuals exhibit as attributable to clumsiness.
Contortionists (such as sideshow Indian rubber man Opens in new window) generally have a form of this disorder. Niccolò Paganini Opens in new window, whose remarkable dexterity and virtuosity on the violin has been attributed by some to an unnatural (dis)ability wrought by Marfan syndrome Opens in new window, has been suggested by others to have instead had EDS.
There is no accepted method of prenatal diagnosis for all forms of EDS, though theoretically Amniocentesis and Chorionic Villus Sampling can successfully detect those forms associated with identified genetic or biochemical defects.
Therapy: Preventive medical needs
Knowledge that a pregnant woman is affected with Ehlers-Danlos syndromes types I-III allows planning for the possibility of maternal bleeding from episiotomy wounds or lacerations, and observation for bladder or rectal prolapse.
There will be a 50% risk of the infant being affected, with a higher risk for rupture of membrane and prematurity. After birth, the infant should be evaluated for signs of joint laxity and skin elasticity, with ophthalmologic and cardiologic consultation in suspect patients.
For the majority of patients with joint hypermobility with or without skin changes, counseling to limit high intensity/collision sports and monitoring of joint and skeletal status will then be sufficient.
However, the potential for serious cardiovascular complications should be borne in mind, with ready referral to cardiology or gastroenterologists for cardiopulmonary symptoms or intestinal/abdominal complaints. If EDS type IV is suspected based on severe early bruising in the children, then early and annual cardiology assessments are essential.
Dental care is even more important in these children, and the potential for scoliosis (spinal curvature) and joint dislocation make a baseline orthopedic evaluation worth considering once the child begins walking.
In the adolescent and adult years, surveillance of the eyes, skin, and joints should continue with auscultation for signs of heart disease. Patients who have had an earlier echocardiogram to rule out congenital heart lesions can probably be followed asymptomatically, but changing auscultatory or clinical signs warrant repeat echocardiography.
Bracing fusions seem to be the most common methods for orthopedic treatment of injured joints, including spinal fusions for severe curvatures. Cosmetic surgery and/or physical therapy/ trainer consultation regarding alternative activities may be worthwhile for adolescents and young adults to promote a healthy lifestyle and body image.
- Abel MD, Carrasco LR: Ehlers-Danlos syndrome: classifications, oral manifestations, and dental considerations, Oral Surg Oral Med Oral Pathol Oral Radiol Endod 101:582-590,2006.
- Byers PH, Murray ML: Heritable collagen disorders: the paradigm of the Ehlers-Danlos syndrome, J Invest Dermatol 132(E1):E6-E11, 2012.
- Byers PH, Murray ML: Ehlers-Danlos syndrome: A showcase of conditions that lead to understanding matrix biology,Matrix Biol 33:10-15, 2014.
- De Coster PJ, Malfait F, Martens LC, et all: Unusual oral findings in dermatosparaxis (Ehlers-Danlos type VIIC), J Oral Pathol Med 32:568-570, 2003.
- De Paepe A, Malfait F: The Ehlers-Danlos syndrome, a disorder with many faces, Clin Genet 82:1-11, 2012.
- Pepin M, Schwarze U, Superti-Furga A, et al: Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type, N Engl J Med 342:673-680, 2000.
