
Griscelli Syndrome Type 2 (GS with Hemophagocytic Syndrome)
Clinical Features and Genetic Patterns
|
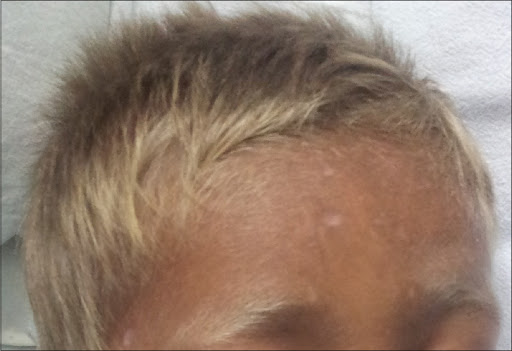
The Griscelli syndrome type 2 (GS2; OMIM #607624) is characterized by hypomelanosis with immunologic abnormalities with or without neurological impairment. The GS2 phenotype currently corresponds to the original patients reported by Griscelli et al. (1978). GS2 patients exhibit various degrees of skin hypopigmentation Opens in new window and a silvery-gray sheen of the hair with large pigment aggregates in hair shafts. |
In most patients at least one episode of hemophagocytic syndrome (HS) Opens in new window or hemophagocytic lymphohistiocytosis (HLH) Opens in new window (the so-called accelerated phase), which is a lymphohistiocytic proliferation of unknown origin consisting of:
- multivisceral infiltration,
- hemophagocytosis,
- pancytopenia,
- hypertriglyceridemia,
- hypofibrinogenemia, and
- hypoproteinemia.
It is triggered by infectious episodes (usually viral but also bacterial) and is associated with a poor prognosis.
When a remission is obtained, recurrent, accelerated phases with increasing severity are seen.
The immunodeficiency is characterized by absent delayed-type cutaneous hypersensitivity and impaired natural killer cell function. No abnormal cytoplasmic granules are present in leukocytes.
Patients with GS2 have immunologic abnormalities during the course of the hemophagocytic syndrome leading to leukocyte brain infiltration that sometimes result in secondary neurological involvement with diffuse white matter abnormalities seen at MRI.
Main signs include hyperreflexia, seizures Opens in new window, signs of intracranial hypertension, (e.g., vomiting or altered consciousness), strabismus, dysatrhia, ataxia, or regression of developmental milestones.
The primary clinical differentiation between GS2 and GS1 Opens in new window is that the former has no primary neurological features. Occasionally, neurological problems may be first sign of the accelerated phase.
CT and MRI Diagnostic
CT and MRI findings are usually normal at birth. When the disease manifests, imaging findings are normal: CT can show areas of coarse calcification in the globus pallidus bilaterally, left parietal white matter, periventricular and left brachium pontis.
Patients with GS2 can manifest unilateral hypodense signals in the genu and posterior limb of the internal capsule (compatible with inflammatory changes), as well as a posterior aspects of both thalami, together with minimal generalized atrophy.
CT scanning can also suggest cell infiltration of the brain. The subcortical white matter can be affected as occurs in the GS1 variant.
Cause: Gene Description
GS2 is caused by mutations Opens in new window in the RAB27A gene which encodes a GTP-binding protein (rab27) that functions in the targeting and fusion of transport vesicles with their appropriate acceptor membranes.
Like other rab proteins, rab27 requires geranylgenarylation of two consensus C-terminal cysteine residues in order to be anchored to membranes.
Truncation of the carboxy-terminal part of rab27 would render it inactive. To date all patients with GS and mutations in RAB27A have developed the hemophagocytic syndrome.
See also:
- Aksu G, Kutekculer N, Genel F, Vergin C, Omowaire B (2003) Griscelli syndrome without hemophagocytosis in an eleven-year-old girl: expanding the phenotypic spectrum of Rab27A mutations in humans. Am J Med Genet A 116:329-333.
- Anikster Y, Huizing M, Anderson PD, Fitzpatrick DL, Klar A, Gross-Kieselstein E, Berkun Y, Shazberg G, Gahl WA, Hurvitz H (2002) Evidence that Griscelli syndrome with neurological involvement is caused by mutations in Rab27A, not Myo5a. Am J Hum Genet 71:407-414.
- Arico M, Xecca M, Santoro N, Caselli D, Maccario R, Danesino C, de Saint Basile G, Locatelli F (2002) Successful treatment of Griscelli syndrome with unrelated donor allogenic hematopoietic stem cell transplantation. Bone Marrow Transplant 29: 995-998.
- Bahadoran P, Ortonne JP, Ballotti R, de Saint-Basile G (2003b) Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet 166A:408-409.
- Elejalde BR, Holguin J, Valencia A, Gilbert EF, Molina J, Marin G, Arango LA (1979) Mutations affecting pigmentation in man: I. Neuroectodermal melanosomal disease. Am J Med Genet 3: 65-80.
- Fukuda M (2005) Versatile role of Rab27 in membrane trafficking: focus on the Rab27 effector families. J Biochem 137:9-16.
- Griscelli C, Durandy A, Guy-Grand D, Daguillard F, Herzog C, Pruneiras M (1978) A syndrome associating partial albinism and immunodeficiency. Am J Med 65: 691-702.
- Huizing M, Anikster Y, Gahl WA (2002) Reply to Menasche et al. Am J Hum Genet 71:1238.
- Ivanovich J, Mallory S, Storer T, Ciske D, Ciske D, Hing A (2001) 12-year-old male with Elejalde syndrome (neuroectodermal melanolysomal disease). Am J Med Genet 98:313-316.
- Mamishi S, Modarressi MH, Pourakbari B, Tamizifar B, Mahjoub F, Fahimzad A, Alyasin S, Bemanian MH, Hamidiyeh AA, Fazlollahi MR, Ashrafi MR, Iaseian A, Khotaei G, Yeganeh M, Parvaneh N (2008) Analysis of RAB27A Gene in Griscelli Syndrome type 2: Novel Mutations Including a Deletion Hotspot. J Clin Immunol Mar 19.
- Mancini AJ, Chan LS, Paller AS (1998) Partial albinism with immunodeficiency: Griscelli syndrome. Report of a case and review of the literature. J Am Acad Dermatol 38:295-300.
- Manglani M, Adhvaryu K, Seth B (2004) Griscelli syndrome – a case report. Indian Pediatr 41:734-737.
- Menasche G, Pastural E, Feldman J, Certain S, Ersoy F, Dupuis S, Wulfrant N, Bianci D, Fisher A, Le Deist F, de Saint Basile G (2000) Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet 25: 173-176.
- Menasche G, Fisher A, de Saint Basile G (2002) Griscelli syndrome types 1 and 2. Am J Hum Genet 71: 1237-1238.
- Menasche G, HoCh, Sanal O, Feldmann J, Tezcan I, Ersoy F, Houdusse A, Fischer A, de Saint Basile G (2003) Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest 112: 450-456.
- Elejalde BR, Valencia A, Gilbert EF, Marin G, Molina J, Holguin J (1977) Neuro-ectodermal melanolysosomal disease: an autosomal recessive pigment mutation in man. Am J Hum Genet 29:39A (abstract).
