
Acromegaly
Clinical Definition
Acromegaly is a syndrome of bony and soft tissue overgrowth that results from excessive circulating growth hormone (GH) occurring after puberty. If the GH excess occurs before epiphysial closure, tall stature and gigantism result.
The cause of this hormonal excess in the vast majority of cases is a GH-secreting pituitary adenoma, frequently with tumor size >1 cm (macroadenoma Opens in new window).
The metabolic effects of the GH are mediated in large part by release of insulin-like growth factor I (IGF-I) from the liver and other tissues.
Clinical Manifestations
The main signs and symptoms of acromegaly include:
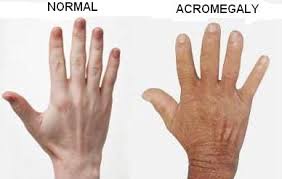
- enlargement of the hands, feet, jaw, and skull
- abnormal overgrowth of internal organs
- patients may notice increasing glove and ring size caused by hand enlargement and finger widening.
Cutaneous abnormalities
Cutaneous changes in acromegaly include hyperphydrosis and oily skin. The GH-indcued changes also give the hands a distinctive doughy, moist character that can be detected on handshake.
Associated carpal tunnel syndrome Opens in new window is common. Increasing foot width may necessitate changing show size.
Growth of the mandible causes protrusion of the jaw (prgnathism) and dental malocclusion. Enlargement of the bones of the skull and paranasal sinuses may alter facial appearance over time and result in increased hat size.
Other abnormalities
Pharyngeal overgrowth may result in obstructive sleep apnea Opens in new window, and laryngeal enlargement produces a characteristic deepening of the voice.
Left ventricular hypertrophy Opens in new window, cardiomegaly Opens in new window, and frequently associated hypertension contribute to increased incidence of congestive heart failure and other cardiovascular mobidity.
Arthritis and arthralgias are common in acromegaly, especially affecting the large joints (hips and shoulders). Impaired carbohydrate tolerance and hypogonadism are also frequently seen in acromegaly.
Several centers have noted a significant increase in adenomatous colonic polyps and colorectal cancer in acromegalic patients and recommend surveillance colonoscopy, although this association has been recently challenged by others.
Diagnostic Examination
Biochemical testing to confirm the diagnosis of acromegaly includes the demonstration of fasting elevations in IGF-I and a failure of GH suppression after an oral glucose challenge.
Associated findings may include fasting hyperglycemia and elevations in serum prolactin or phosphorus. The diagnosis of acromegaly is associated with a pituitary mass lesion demonstrable by MRI in more than 90% of patients.
Gene: Dysfunction of G Protein-Regulated Pathways in Acromegaly
Hypothalamic GH-releasing hormone (GHRH) acting through a receptor coupled to Gs and utilizing cAMP as a second messenger normally regulates release of GH from pituitary somatrophs.
Studying the in vitro properties of GH-secreting adenomas removed from patients with acromegaly, Vallar et al. identified a subset of tumors (group 2) with increased basal GH secretion and elevated intracellular cAMP levels that did not respond further to GHRH treatment (Vallar et al., 1987).
The remaining GH-secreting tumors (group 1) had low levels of basal cAMP and GH secretion but showed significant stimulation by GHRH treatment.
The presence of a constitutively activated Gs regulatory protein in the group 2 tumors was inferred from these biochemical studies, and indeed subsequent investigation found activating missence mutations in tumor-derived Gsα cDNA from four of four group 2 tumors (Landis et al., 1989).
These mutations encoded Arg201 →Cys (R201C), Arg201 →His (R201H), and Gin227 →Arg (Q227R) changes in Gsα leading to its constitutive activation.
Series of GH-secreting adenomas analyzed in various centers demonstrated mutations in residues 201 or 227 of Gsα in 4% to 53% of tumors. As noted above, acromegaly associated with Arg201 mutations in Gsα can be a component of MAS.
The mechanism of activation of Gsα by mutation of Arg201 and Gin227 has been inferred from structural and biochemical studies to result from loss of GTPase activity.
The structure of Gsα, like that of Giα and Gtα, consists of an α-helical domain bound through two linking peptides to a p21 Ras-like domain containing a six-stranded β-sheet.
The Ras-like domain contains the high-affinity guanine nucleotide-binding site. Three flexible switch elements (switches I–III) in Gα subunits adopt different conformations in the Gα-GDP vs. Gα-GTP states and provide surfaces for conformation-dependent interaction of Gα with Gβγ complexes, RGS proteins, and effector molecules.
Switch I corresponds to one of the two linker peptides joining the Gα helical domain with the Ras-like domain and contains Arg201 in Gsα.
This arginine residue is thought to facilitate GTP hydrolysis by stabilizing a transition-state pentavalent phosphate and is the same arginine in Gsα covalently modified by cholera toxin resulting in constitutive signal activation. Mutation of this residue in vitro leads to diminished GTPase activity and constitutive activation of Gsα.
The switch II region in the Ras-like domain of Gsα contains GIn227, which corresponds to Gin61 in p21-ras, site of GTPase-inactivating oncogenic point mutations.
The role of this glutamine in GTP hydrolysis is not fully understood, but structural studies suggest it may stabilize the orientation of a catalytic water molecule.
Mutation of this glutamine in Gsα can be shown in vitro to inhibit GTPase activity and constitutively activate AC.
Treatment of Acromegaly
The goal of treatment in acromegaly is to reduce GH levels to <1 ng/ml 2 hours after a standard oral glucose load (75 g) and to normalize IGF-I levels relative to age- and sex-matched controls.
Surgical, radiological, and medical treatment modalities directed at GH-secreting pituitary adenomas are available.
- Surgery
Transsphenoidal pituitary microsurgery is the preferred initial treatment for most patients with acromegaly.
As preoperative imaging and neurosurgical techniques have improved, the rate of successful pituitary surgery in acromegaly has risen significantly.
In a study with a mean follow-up period of 16 years, nearly 20% of acromegalic patients developed recurrence after initially successful surgery, whereas 40% of patients treated with surgery only remained cured of their disease.
Poor prognostic factors for surgical cure in acromegaly include macroadenomas >20 mm and preoperative GH levels >50 ng/ml.
- Radiotherapy
Radiotherapy plays an important adjunctive role in the treatment of acromegaly, especially in cases of large or invasive tumors, although there is typically a long delay between treatment and GH response. Partial or complete hypopituitarism is a common late sequel to pituitary irradiation for acromegaly.
- Pharmacology
Pharmacologic treatment of acromegaly with D2 dopamine receptor agonists has shown mixed results. The parenteral administration of long-acting somatostatin analogs has been shown to reduce GH and IGF-I levels and to shrink tumors in previously untreated acromegalic patients. Preliminary studies with the novel GH antagonist pegvisomant appear promising.
See also:
- Ford ZK, Dourson AJ, Liu X, Lu P, Green KJ, Hudgins RC, et al. Systemic growth hormone deficiency causes mechanical and therma hypersensitivity during early postnatal development. IBRO Reports. 2019;6:111-121
- Tritos NA. Focus on growth hormone deficiency and bone in adults. Best Practice & Research Clinical Endocrinology & Metabolism. 2017;31:49-57
- Vestergaard P, Jorgensen JO, Hagen C, et al. Fracture risk is increased in patients with GH deficiency or untreated prolactinomasea case-control study. Clinical Endocrinology. 2002;56:159-167
- Wuster C, Abs R, Bengstsson BA, et al. The influence of growth hormone deficiency, growth hormone replacement therapy, and other aspects of hypopituitarism on fracture rate and bone mineral density. Journal of Bone and Mineral Research. 2001;16:398-405
- Beshyah SA, Freemantle C, Thomas E, et al. Abnormal body composition and reduced bone mass in growth hormone deficient hypopituitary adults. Clinical Endocrinology. 1995;42:179-189. DOI: 10.1111/j.1365-2265.1995.tb01860.x
