
Neurofibromatosis
Definition, Epidemiology and Clinical Features
|
Neurofibromatosis is an autosomal dominant neurocutaneous syndrome with clinical manifestation associated with difficulty in seeing, hearing, and in some cases with paralysis and early death. Neurofibromatosis belongs in the category of phakomatosis Opens in new window, one of a group of related conditions, hereditary in origin, with glial overgrowth and proliferation or malformations involving the brain and retina. |
Neurofibromatosis comprises two genetically distinct forms: neurofibromatosis type 1(NF1) Opens in new window also known as von Recklinghausen, which was first described in 1882 by the German pathologist, Friedrich Daniel von Recklinghausen (thus the origin of the name Von Recklinghausen’s disease), and neurofibromatosis type 2 (NF2) Opens in new window, characterized by cranial nerve VIII tumors.
In addition to the two subtypes, is a third subtype, neurofibromatosis type 3 (NF3), which is associated with the occurrence of schwannomas on other cranial and spinal nerves apart from the eight cranial nerve commonly described as schwannomatosis.
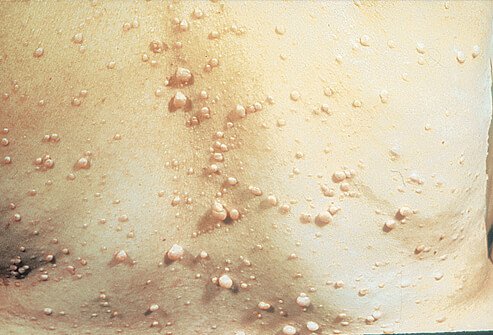
Historically, neurofibromatosis type 1 was considered a peripheral disease with skin manifestations such as café-au-lait or café noir in white and black populations respectively, Lisch nodules, axillary freckling and peripheral neurofibromas.
The current consensus is that, in addition to the skin lesions, patients have central nervous system lesions such as optic nerve gliomas, and other tumors including neuroblastomas.
The occurrence of neurofibromatosis as an autosomal dominant inheritance Opens in new window is 1 in 4000 persons and 1 in 50 000 persons for NF1 and NF2, respectively. The clinical features and manifestations of each type are extremely variable.
Unlike other neurocutaneous syndromes, seizures occur in approximately 10–15% of patients with neurofibromatosis and it is exceptional to patients with intracranial lesions that occur in the cerebral cortices and thalami. It is of particular importance to state that, of the two disease entities, NF1 is more frequently associated with seizures Opens in new window and epilepsy compared to NF2.
Neurofibromatosis type 1 is caused by mutations in the 61-exon gene on chromosome 17q. Clinical features include multiple fleshy skin tags and deeper soft tissue tumors (neurofibromas), ‘café-au-lait’ spots (light-brown macules of varying size), axillary freckling, scoliosis, and an increased incidence of a variety of neural tumors, e.g., meningioma, eighth-nerve tumors and gliomas.
Neurofibromatosis type 2 is caused by mutations in the NF2 gene (encoding for the protein Merlin or Schwannomin) on chromosome 22 p and presents with bilateral acoustic neuromas, other neural tumors and cutaneous neurofibromas.
Owing to the fact that different gene mutations are responsible for the two disease entities, it is unlikely for both NF1 and NF2 to occur in different members of the same family. It must be stated that, regardless of the advances in genetics of neurofibromatosis, wholesome genetic testing for patients with neurofibromatosis is still debated on the premise of cost and the small effect genetic testing has on the clinical care of affected persons.
Epilepsy in the Disease
Typically, focal seizures are prevalent in neurofibromatosis, and almost all types of focal seizures including focal dyscognitive seizures are described.
In one series of 630 patients with neurofibromatosis, 37 patients had epileptic seizures, out of which 17 patients had partial seizures, and a further 14 of these seizure types were simple partial seizures.
Generalized seizure types that were identified in this cohort included absences, GTCSs, and infantile spasms.
In this cohort, once seizures were diagnosed they were difficult to control, with most patients requiring polytherapy, even though previously it was thought to be fairly easy to control, with evidence that up to 60–70% of individuals will achieve seizure freedom on monotherapy or even no antiepileptic medications.
Seizures Opens in new window occur in approximately 15% of patients with neurofibromatosis type 1 and only about 1% of patients with neurofibromatosis type 2 will ever experience seizures. The cause for this disparity remains unclear; however, the involvement of the mTOR pathway in neurofibromatosis type 1 may be a significant reason, as abnormalities in the function of the latter have been associated with the development of epileptic seizures in animal models of epilepsy.
Macrocephaly Opens in new window is reported to occur in 10% of neurofibromatosis patients, which may be associated with the development of seizures. Mental retardation Opens in new window, learning disabilities and hyperactivity are described in the clinical features of neurofibromatosis. Though not necessarily related, intellectual disability and seizures occur in 10% of patients with neurofibromatosis.
Unlike tuberous sclerosis Opens in new window, particular TSC2 gene mutations, where a reason for a common pathway to seizure development and the intellectual disability is suspected, no obvious link has been identified in neurofibromatosis.
Hypothalamic gliomas and/or hamartomas are seen in neurofibromatosis predisposing them to gelastic seizures. Continuous seizures including status epilepticus and epilepsia partalis continua are reported in patients with neurofibromatosis.
On the foregoing evidence, it is conceivable that the prevalence of seizures will be high in patients with neurofibromatosis; however, only 3–6% of patients actually have seizures compared with other neurocutaneous syndromes such as tuberous sclerosis. It is believed that the prevalence of seizures increases in neurofibromatosis when the genetic abnormality is maternally inherited and this has been attributed to a reduced fitness in male patients with neurofibromatosis, although there are no current studies with evidence to support this claim.
It follows from the preceding discussion that neurofibromatosis is a cause of epilepsy, has a complex epilepsy phenotype, and may be described as epilepsy associated with single gene disorders.
The prevalence of epilepsy in neurofibromatosis, regardless of the numerous cerebral lesions, genetic anomalies and the risk of development of seizures and thus epilepsy, is particularly low when compared with other neurocutaneous syndromes.
On the other hand, the risk of developing epilepsy in neurofibromatosis is lifelong; males and females are equally affected and no age-specific incidence has been identified. There is generally a higher prevalence of seizures in epilepsies in neurofibromatosis. The epilepsies in neurofibromatosis are correctly classified as symptomatic epilepsies — predominately of genetic or developmental causation according to the currently accepted listing of the cause of epilepsy, and are mostly focal epilepsies.
When cognitive decline presents in patients with neurofibromatosis, the onset of epilepsy in such patients is associated with significant increase in the total disease burden, which may be further compromised by the introduction of antiepileptic medication.
Diagnosis of Neurofibromatosis
Clinical diagnosis of neurofibromatosis 1 can be made with a high degree of accuracy when at least two of the following features are present.
- Six or more café-au-lait macules (or café noir in the black population) > 5 mm in greatest diameter in prepubertal individuals or > 15 mm in greatest diameter in postpubertal individuals.
- Two or more neurofibromas of any type or one plexiform neurofibroma.
- Axillary or inguinal freckling.
- Optic nerve gliomas.
- Two or more Lisch nodules (iris hamartomas).
- A distinctive osseous lesion such as sphenoid dysplasia or tibial pseudo-arthrosis.
- A first-degree relative (parent, sibling, or offspring) with NFI as defined by the above criteria.
Neurofibromatosis type 2 may be diagnosed when a patient presents with:
- Bilateral vestibular schwannomas, or
- First-degree relatives with unilateral schwannomas before the age of 30 years or with any two of the following — neurofibroma, meningioma, gliomas and juvenile subcapsular lenticular opacities.
The diagnosis is usually straightforward using these criteria for clinical evaluation, although in some situations expert histological review of tumors as well as expert radiological review of neuroimaging may be necessary. The commonest presentation in a child is multiple café-au-lait (café noir in the black population) spots.
Differential diagnosis of café-au-lait spots Opens in new window includes various ring chromosome syndromes, but in the case of ring chromosomal syndromes Opens in new window the child will usually have more developmental complications as well as dysmorphic features than is realized in neurofibromatosis type 1 Opens in new window.
In cases where there is varied pigmentation of the skin lesions and the variation is in the intensity of the pigmentation with significant irregularity of the edges, or the involvement of very large segments of the body, other diagnoses such as DNA repair syndromes (Legius syndrome and constitutional mismatch repair deficiency [CMMRD] syndrome Opens in new window), McCune-Albright syndrome Opens in new window or pigmentary miscegeny (where parents have very different skin coloring) needs to be considered.
Affected persons with Legius syndrome Opens in new window have café-au-lait spots Opens in new window and axillary freckling indistinguishable from NF1 Opens in new window; however, the clinical features differentiating NF1 and Legius syndrome are the absence of Lisch nodules and neurofibromas.
Management of Epilepsy in Patients with Neurofibromatosis
Epilepsy occurs in 6–7% of patients with neurofibromatosis. Neurological examination should be undertaken annually during routine medical check-up. Neurological complications including epilepsy will develop from tumors and cortical malformations, and any unexplained neurological signs and symptoms should necessitate a referral to the neurologist.
Specialist multidisciplinary clinics with focus on neurofibromatosis are encouraged. The roles of these clinics are to identify difficult cases, and educate and support the family, including provision of genetic counseling. The latter is advised for all patients with neurofibromatosis.
When the first child of unaffected parents has neurofibromatosis, it is essential to examine and search for cutaneous lesions and/or Lisch nodules in the parent; if these are absent, the risk of neurofibromatosis in subsequent children will be very low and has been described to be less than 1%.
Individuals with the NF1 gene mutation have a 50% chance of having offspring who are affected; however, the severity of the clinical problems cannot be determined. There is currently no known cure for neurofibromatosis.
Recent data suggest the possibility of use of anti-vascular endothelial growth factor (VEGF) antibody, bevacizumab, in patients with neurofibromatosis type 2.
Annual follow-up with a medical doctor familiar with this condition is suggested, and in most cases a multidisciplinary approach to treatment is recommended with referrals to ophthalmologists, cardiologists, and spine and orthopedic surgeons for the treatment and management of related complications.
Treatment of epilepsy in neurofibromatosis is adapted to the affected individual. Routine EEG for all patients with neurofibromatosis is suggested; however, the yield and specificity is low, making the diagnosis more clinical.
Pattern abnormalities on EEG include focal spike discharges, multifocal discharges, and 3 Hz spike wave discharges. Seizures usually remain nonthreatening and highly sensitive to antiepileptic medications and are readily controlled on standard anticonvulsant medications; most patients will become free of seizures on one anticonvulsant medication.
At times no medication may be required. This is in contrast to other neurocutaneous syndromes such as tuberous sclerosis where antiepileptic polytherapy may be required. It is stated that no one anticonvulsant confers a singular advantage in seizure control in neurofibromatosis.
Epilepsy surgery is limited to the few cases that have neuroimaging evidence of malformations of cortical development and/or glioneuronal tumors. These patients tend to develop intractable seizures. Provided there is single well-defined epileptogenic lesion during preoperative assessment, surgery can be successfully done in patients with neurofibromatosis.
The combined prognosis for the control of epilepsy in neurofibromatosis is very comparable to the general population and remains much better than what is found in other neurocutaneous syndromes.
See also:
- Kehrer-Sawatzki H, Farschtschi S, Mautner VF, Cooper DN, (2017) The molecular pathogenesis of schwannomatosis, a paradigm for the co-involvement of multiple tumor suppressor genes in tumorigenesis. Human Genetics 136(2):129–48.
- National Institutes of Health Consensus Development Conference Statement: neurofibromatosis. Bethesda, Md., USA, July 13–15, 1987. Neurofibromatosis 1988;1(3):172-8.
- Riccardi VM. (1992) The prenatal diagnosis of NF-1 and NF-2. The Journal of Dermatology 19(11):885–91.
- Creange A, Zeller J, Rostaing-Rigattieri S, et al. (1999) Neurological complications of neurofibromatosis type 1 in adulthood. Brain: a Journal of Neurology 122(Pt 3):473–81.
- DiFrancesco JC, Sestini R, Cossu F, et al. (2014) Novel neurofibromatosis type 2 mutation presenting with status epilepticus. Epileptic Disorders: International Epilepsy Journal with Videotape 16(1):132–7.
- Ribierre T, Baulac S. (2017) mTOR pathway in familial focal epilepsies. Oncotarget 8(4):5674-5.
- Caban C, Khan N, Hasbani DM, Crino PB. (2017) Genetics of tuberous sclerosis complex: implications for clinical practice. The Application of Clinical Genetics 10:1–8.
- Tanito K, Ota A, Kamide R, Nakagawa H, Niimmura M. (2014) Clinical features of 58 Japanese patients with mosaic neurofibromatosis 1. The Journal of Dermatology 41(8):724–8.
- Easton DF, Ponder MA, Huson SM, Ponder BA. (1993) An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): evidence for modifying genes. American Journal of Human Genetics 53(2):305–13.
- Morrow KA, Shevede LA. (2012) Merlin: the wizard requires protein stability to function as a tumor suppressor. Biochimica et Biophysica Acta 1826(2):400–6.
- Ruggieri M, Pratico AD, Serra A, et al. (2016) Childhood neurofibromatosis type 2 (NF2) and related disorders: from bench to bedside and biologically targeted therapies. Acta Otorhinolaryngologica Italica 36(5): 345–67.
- Hsieh HY, Fung HC, Wang CJ, Chin SC, Wu T. (2011) Epileptic seizures in neurofibromatosis type 1 are related to intracranial tumors but not to neurofibromatosis bright objects. Seizure 20(8):606–11.
- Vivarelli R, Grosso S, Calabrese F, et al. (2003) Epilepsy in neurofibromatosis 1. Journal of Child Neurolog 18(5):338–42.
- Ostendorf AP, Gutmann DH, Weisenberg JL. (2013) Epilepsy in individuals with neurofibromatosis type 1. Epilepsia 54(10):1810–14.
- Korf BR, Carrazana E, Holmes GL. (1993) Patterns of seizures observed in association with neurofibromatosis 1. Epilepsia 34(4):616-20.
- Kulkantrakorn K, Geller TJ. (1998) Seizures in neurofibromatosis 1. Pediatric Neurology 19(5):347-50.
- Ruggieri M, Mastrangelo M, Spalice A, et al. (2011) Bilateral (opercular and paracentral lobular) polymicrogyria and neurofibromatosis type 1. American Journal of Medical Genetics Part A 155a(3): 582-5.
- Okazaki K, Kakita A, Tanaka H, et al. (2010) Widespread ischemic brain lesions caused by vasculopathy associated with neurofibromatosis type 1. Neuropathology: Official Journal of the Japanese Society of Neuropathology 30(6):627-33.
- Huson SM, Harper PS, Compston DA. (1998) Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain: a Journal of Neurology 111(Pt 6): 1355-81.
- Gales J, Prayson RA. (2016) Hippocampal sclerosis and associated focal cortical dysplasia-related epilepsy in neurofibromatosis type 1. Journal of the Neurosurgical Society of Australasia 37:15-19.
- Friedman JM, Birch PH. (1997) Type 1 neurofibromatosis: a descriptive analysis of the disorder in 1,728 patients. American Journal of Medical Genetics 70(2):138-43.
- Rauen KA, Huson SM, Burkitt-Wright E, et al. (2015) Recent developments in neurofibromatoses and RASopathies: management, diagnosis and current and future therapeutic avenues. American Journal of Medical Genetics Part A 167(1):1-10.
- Plotkin SR, Bredella MA, Cai W, et al. (2012) Quantitative assessment of whole-body tumor burden in adult patients with neurofibromatosis. PLoS ONE 7 (4): e35711.
- Ferner RE, Huson SM, Thomas N, et al. (2007) Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. Journal of Medical Genetics 44(2):81-8.
- Subbiah V, Slopis J, Hong DS, et al. (2012) Treatment of patients with advanced neurofibromatosis type 2 with novel molecularly targeted therapies: from bench to bedside. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology 30(5):e64-8.
