
Osteogenesis Imperfecta
Clinical Features of Type III Osteogenesis Imperfecta
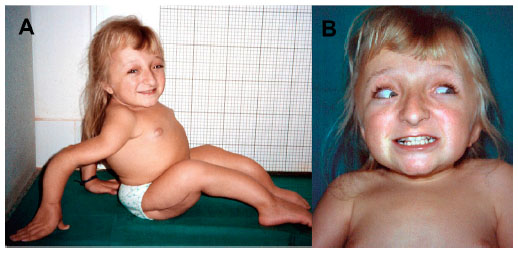 Figure X-1, Clinical manifestations seen in a patient with Type III OI | Photo courtesy of Dovepress Opens in new window
Figure X-1, Clinical manifestations seen in a patient with Type III OI | Photo courtesy of Dovepress Opens in new window
Type III form of osteogenesis imperfecta Opens in new window is a rare autosomal recessive disorder characterized by moderate to severe bone fragility.
Intrauterine fractures are characteristic along with bone deformity, short stature, and triangular facies with frontal bossing and pulmonary hypertension. These patients usually require multiple orthopedic procedures throughout life.
Other associated features of Type III osteogenesis imperfecta include dentinogenesis imperfecta Opens in new window, sclera of variable hue, limb shortening and progressive deformities.
In utero fractures occur in 50% of cases. The remaining half of cases have fractures in the neonatal period. No hearing loss has been reported in this type.
These clinical findings and the likes are elaborated within the remainder of this entry.
- General features
This osteogenesis imperfecta syndrome, characterized as progressively deforming with normal sclera, is usually not lethal, at least in the newborn period.
Beighton and Versfeld and Viljoen and Beighton have reported a high prevalence of an autosomal recessive form among the black population in South Africa.
The diagnosis in sporadic cases with an osteogenesis imperfecta type III phenotype is confounded by clinical and radiographic heterogeneity, and by clinical similarity to other patients with severe osteogenesis imperfecta Opens in new window.
The extent of this heterogeneity is unknown; Hanscom and Bloom recognized four subtypes.
In a recent study approximately one-third of patients survived long term, reflecting not only the severity of the disorder but also heterogeneity within the group. Of 17 patients with the disorder, 4 died in the first year and 5 in the second and third decades.
Death usually results from complications of severe bone fragility, skeletal deformity including kyphoscoliosis, pulmonary hypertension, and cardiopulmonary failure.
Thompson et al calculated an empirical recurrence risk of 7% for families of children who have severe osteogensis imperfect but who survive the perinatal period.
They concluded that about 75% represented new autosomal dominant mutations, whereas 25% were autosomal recessive.
In a black population originating from South Africa, the chance for an autosomal recessive form was much higher. Both autosomal recessive inheritance and gonadal mosaicism for type III have been demonstrated molecularly.
- Ophthalmologic abnormalities
Although blue sclera are observed in infancy, the blueness fades by 1 year of age, so that older patients have pale blue or white sclera.
- Otolaryngologic abnormalities
Although hearing loss has been said to occur infrequently, audiologic findings have not been well documented in patients with unequivocal osteogenesis imperfecta type III.
- Neurologic abnormalities
On head CT scans, diffuse ventricular dilatation and diffuse cortical atrophy have been described. There is one report of a child with osteogenesis imperfecta type III and an arachnoid cyst Opens in new window.
No associated increase in intracranial pressure, cranial nerve dysfunction, or herniation syndrome was noted.
- Cardiovascular system
Significant cardiac or vascular lesions have not been described, but asymptomatic mitral valve prolapse and type II atrial septal defects have been found.
- Joint abnormalities
Ligamentous laxity is marked in children but is less severe in adults.
- Skeletal findings
Infants with this syndrome are usually born at or near term with normal birth weight; birth length is usually normal, but some have marked deformity. With age, all fall well below the third centile in height for age and sex.
Head size is disproportionately large compared to the rest of the body. The ossification defect in the skull is not as severe as in osteogenesis imperfecta type II Opens in new window.
Frontal and temporal bossing contribute to the triangular facies. The anterior fontanel, sutures, and posterior fontanel are wide, and wormian bones or bone islands may be palpable along the posterior sutures.
Fractures are present at birth in half of these infants; all have numerous fractures by 1 or 2 years of age. Although multiple rib fractures occur, continuous beading as seen in osteogenesis imperfecta type II Opens in new window is not found.
Long bones are also subject to multiple fractures and bowing and, in some cases, there is metaphyseal flaring.
The lower extremities are often already bowed at birth. The limbs are not as short or as deformed as in osteogenesis imperfecta type II. Some femora are normal, whereas others are short and broad.
Femurs and tibias may be markedly angulated. In some patients, neonatal radiographs demonstrate broad metaphyses with centrally overmodeled diaphyses and angulation deformities; during the first year of life, diaphyses broaden.
Sillence et al demonstrated in a longitudinal study that many patients had marked thickening of the femoral shafts during the first few years of life; this morphology makes it difficult to distinguish these patients from those with osteogenesis imperfecta type IIB. With time, however, progressive narrowing occurs, so that in older patients, femurs are thin. Protrusio acetabulum is often severe.
In the first few years, metaphyses develop increasing density and irregularity, which progress so that by the end of the first decade, metaphyses and epiphyseal zones are replaced by whorls of radiodensity, giving a cystic appearance.
Progressive and marked vertebral flattening is common. Severe kyphoscoliosis also develops.
Most patients become markedly handicapped. Bowing and angulation deformities are likely to be progressive.
- Oral abnormalities
Dental abnormalities similar to those found in the other osteogenesis imperfecta syndromes may be present, but complete radiographic and morphologic evaluation of the teeth has been reported for few patients.
The patient described by Nicholls et al and Pope et al had normal teeth. Within families, some patients have normal teeth, whereas others have teeth that are opalescent.
Lund et al, using in vitro protein-chemical features and molecular mutations for classification, found that 81% of type III patients had dentinogenesis imperfecta.
See also:
- Clinical Features of Type I Osteogenesis ImperfectaOpens in new window
- Clinical Features of Type II Osteogenesis ImperfectaOpens in new window
- Clinical Features of Type IV Osteogenesis ImperfectaOpens in new window
- Osteogenesis Imperfecta (Molecular Pathogenesis of the syndrome)Opens in new window
- Therapeutics for Patients with Osteogenesis ImperfectaOpens in new window
- Cole WG, Lam TP: Arachnoid cyst and chronic subdural hematoma in a child with OI III resulting from the gly 1006 ala substitution in the α2(I) chain of type I procollagen. J Med Genet 33:193-196, 1996.
- Cohen-Solal L et al: Dominant mutations in familial lethal and severe osteogenesis imperfecta. Nature 304:78-80, 1983.
- De Paepe A et al: Homozygosity by descent for a COL1A2 mutation in two sibs with severe osteogenesis imperfecta and mild clinical expression in the heterozygotes. Hum Genet 99:478-483, 1997.
- Vetter U et al: Osteogenesis imperfect in childhood: Cardiac and renal manifestations. Eur J Pediatr 149:184-187, 1989.
- Beighton P, Versfeld GA: On the paradoxically high relative prevalence of osteogenesis imperfecta type III in the black population of South Africa. Clin Genet 27:398-401, 1985.
- Tsipouras P et al: Neurologic correlation of osteogenesis imperfecta. Arch Neurol 43:150-152, 1986.
- Hanscom DA, Bloom BA: The spine in osteogenesis imperfecta. Orthoped Clin North Am 19:449-458, 1988.
- Sillence DO et al: Osteogenesis imperfecta type III. Delineation of the phenotype with reference to genetic heterogeneity. Am J Med Genet 23:821-832, 1986.
- Wallis GA et al: Osteogenesis imperfecta type III: Mutations in type I collagen structural genes, COL1A1 and COL1A2, are not necessarily responsible. J Med Genet 30:492-496, 1993.
- Thompson EM et al: Recurrence risks and prognosis in severe sporadic osteogenesis imperfecta. J Med Genet 24:390-405, 1987.
- Sillence DO et al: Genetic heterogeneity in osteogenesis imperfect. J Med Genet 16:101-116, 1979.
- Verloes A et al: Osteocraniostenosis. J Med Genet 31:772-778, 1994.
- Lund AM et al: Dental manifestations of osteogenesis imperfecta and abnormalities of collagen I metabolism. J Craniofac Genet Dev Biol 18:30-37, 1998.
- Nicholls AC et al: The clinical features of homozygous α2(I) collagen-deficient osteogenesis imperfect. J Med Genet 21:257-262, 1999.
