
Prolidase Deficiency
Clinical Definition and Features
|
Prolidase deficiency is an autosomal recessive inherited Opens in new window inborn error of metabolism characterized by iminodipeptiduria, skin ulcers, mental retardation Opens in new window, characteristic facies, and recurrent infections. |
Normally prolidase cleaves dipeptides containing C-terminal proline or hydroxyproline. When this enzyme Opens in new window is deficient, the normal recycling of proline residues obtained from collagen Opens in new window degradation is impaired.
A build-up of iminodipeptides results, with disturbances in connective tissue metabolism and excretion of large amounts of iminodipeptides Opens in new window in the urine.
In addition, the absence of prolidase activity causes increased keratinocyte apoptosis Opens in new window, which may be in part responsible for the skin lesions.
Cutaneous Manifestations
The disease is inherited as an autosomal recessive disorder Opens in new window and shows variability in age of onset (from 3 months to 17 years) and in severity of clinical expression.
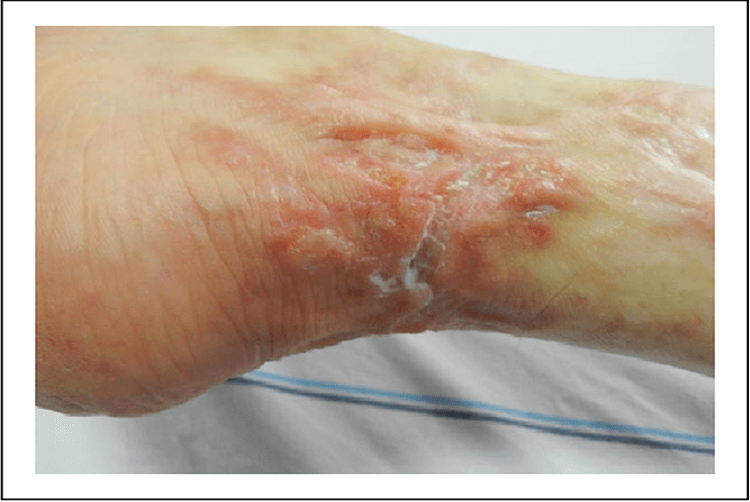
More than 90% of patients with prolidase deficiency have some skin pathology. The most important skin signs, which almost always appear before the affected person is 12 years old, are skin fragility; ulceration and scarring of the lower extremities.
Recurrent multiple ulcers of the lower extremities are the most characteristic clinical findings. Most patients have maculopapular, purpuric, and eczematous eruptions in the first years of life. Leg ulcerations develop near the time of puberty.
Other reported skin findings include telangiectasia Opens in new window, scaly erythematous maculopapular lesions Opens in new window, purpuric lesions Opens in new window, post-inflammatory pigmentation Opens in new window, photosensitivity, and thickening of the skin with lymphedema of the legs.
In addition, five of 25 reported patients have shown premature graying of the hair. The youngest age of onset of gray hair was 7 years. The etiology of the poliosis is not clearly identified.
Extracutaneous Manifestations
Systemic signs and symptoms include mental deficiency; splenomegaly; recurrent infections such as otitis media and sinusistis; hematologic disorders such as anemia Opens in new window and hypergammaglobulinemia Opens in new window.
An unusual facial appearance is noted at times, with low hairline, frontal bossing Opens in new window, and saddle nose Opens in new window. The nasal septum may be perforated; ocular findings, deafness, and short stature have also been reported.
Some patients with prolidase deficiency meet American Rheumatology Association (ARA) criteria for the diagnosis of SLE Opens in new window, but worsen if immunosuppressive treatment is given. Antinuclear antibodies (ANA) and anti ds-DNA may be positive.
Pathogenesis
Prolidase is a proline dipeptidase with strict specificity for substrates containing proline or hydroxyproline at the carboxy terminal end of a peptide. Its gene maps to chromosome 19p13.2 (Ledoux et al., 1994).
Loss of prolidase activity results in accumulation of dipeptides, which inhibits normal recycling of proline and causes alteration in the metabolism of collagen and other proteins that contain a high content of these amino acids.
Prolidase activity has been reported to be deficient in erythrocytes, leukocytes, and cultured skin fibrosis obtained from affected individuals.
Histology
Histologic examination of the skin around the leg ulcers of one patient revealed a deposition of an amorphous substance around the capillaries that stained with Congo red. The collagen fibers showed fragmentation and irregularities in arrangement. No histopathology of the gray hair has been revealed.
Diagnosis
Diagnosis of prolidase deficiency is made by measuring prolidase activity in red blood cells and showing excessive excretion of iminodipeptides in the urine.
Therapeutics
Many therapeutic options have been described, such as oral supplements of manganese and ascorbic acid, both modulators of prolidase activity; however, results of treatment are highly variable across patients.
Topical proline 5% in ointment helped heal chronic leg ulcers in one patient, although it did not prevent the appearance of new ulcerations.
Transfusion of normal erythrocytes have been recommended. Apheresis exchange repeated monthly may improve the leg ulcers. In long standing ulcerations squamous cell carcinomas may occur.
- Leoni, A., G. Cetta, R. Tenni, I. Pasquali-Ronchetti, F. Bertolini, D. Guerra, K. Dyne, and A. Castellani. Prolidase deficiency in two siblings with chronic leg ulcerations: clinical, biochemical and morphologic aspects. Arch. Dermatol. 123:493-499, 1987.
- Milligan, A., R. A. C. Graham-Brown, D. A. Burns, and I. Anderson. Prolidase deficiency: a case report and literature review. Br. J. Dermatol. 121:405-409, 1989.
- Bissonnette, R., D. Friedmann, J. M. Giroux, M. Dolenga, P. Hechtman, V. M. Der Kaloustian, and R. Dubuc. Prolidase deficiency: a multisystemic hereditary disorder. J. Am. Acad. Dermatol. 29:818-821, 1993.
- Goodman, S. I., C. C> Solomons, F. Muschenheim, C. A. McIntyre, B. Miles, and D. O’Brien. A syndrome resembling lathyrism associatied with iminodipeptiduria. Am J. Med. 45:152-159, 1968.
- Arata, J., S. Umemura, Y. Yamamoto, M. Hagiyama, and N. Nohara. Prolidase deficiency: its dermatological manifestations and some additional biochemical studies. Arch. Dermatol. 115:62-67, 1979.
- Freij, B. J., and V. M. Der Kaloustian. Prolidase deficiency: a metabolic disorder presenting with dermatologic signs. Int. J. Dermatol. 25:431-433, 1986.
