
Sickle Cell Anemia
Pathophisiology of Sickle Cell Anemia
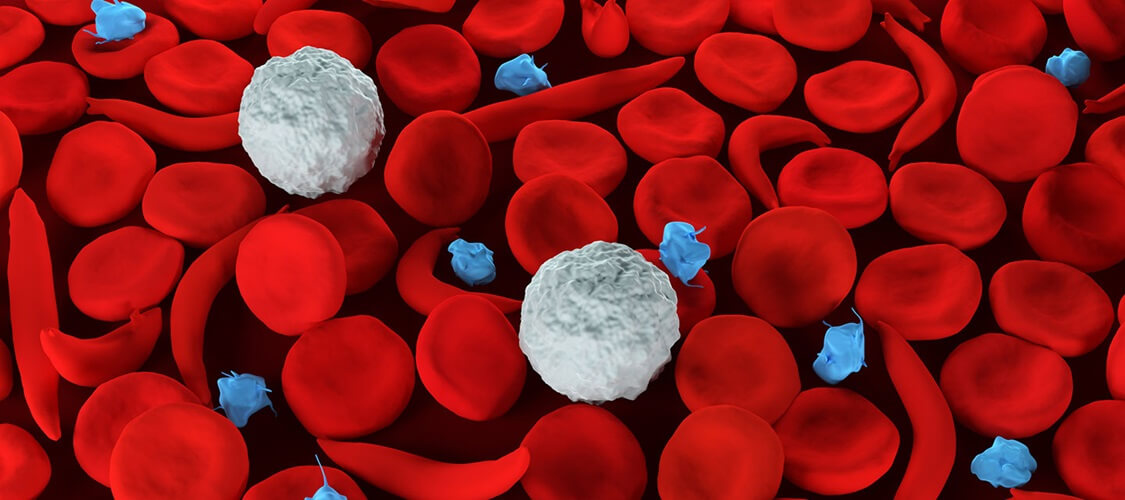
|
Sickle cell anemia is a genetic disease of the blood, caused by a biological defect in one gene of a person. Genes are the elements in cells that carry the information that determines traits, such as hair or eye color. In sickle cell anemia, a defect in the gene controls how hemoglobin is made. This defect can be transmitted from parents to their children. |
One drop of humans’ blood contains millions of red blood cells (RBCs). Red blood cells travel throughout the body to deliver oxygen and take a way waste from tissues. These cells get their color from hemoglobin, which is the chemical in red blood cells that allows them to carry oxygen around the body.
Red blood cells look like doughnuts. They are flat, soft, and round and have a dimple in the middle. They move quickly through tiny blood tubes called capillaries in all parts of the body.
The bodies of people with sickle cell anemia sometimes form red blood cells that look like bananas, or sickles. The long red blood cells are hard and sticky and have pointed ends.
Instead of flowing freely through the capillaries, the sickle cells have the tendency to bunch up. Some break apart. Bunching up or breaking apart slows or stops the flow of blood. Then cells do not get enough oxygen, and waste products from the cells cannot be taken away.
The lack of oxygen and buildup of waste products in the tissues cause severely great pain and can even cause death.
Prevalence
According to the National Institutes of Health (NIH), approximately 72,000 Americans are living with the diseases of sickle cell anemia. Most are African Americans.
Some Hispanic Americans also have the disease. The NIH estimates that millions of people worldwide have sickle cell anemia. It is most common in people who live by large bodies of water in warm areas of the world.
Tests to Detect Sickle Cell Genes Before Birth
Sickle cell disease can be diagnosed in an unborn baby by sampling some of the fluid surrounding the baby in the mother's womb (amniotic fluid). If you or your partner has sickle cell anemia or the sickle cell trait, ask your doctor about this screening.
Treatment
Management of sickle cell anemia is usually aimed at avoiding pain episodes, relieving symptoms and preventing complications. Treatments might include medications and blood transfusions. For some children and teenagers, a stem cell transplant might cure the disease.
- Medications
- Hydroxyurea (Droxia, Hydrea, Siklos). Daily hydroxyurea reduces the frequency of painful crises and might reduce the need for blood transfusions and hospitalizations. It can also increase your risk of infections. Don't take the drug if you're pregnant.
- L-glutamine oral powder (Endari). The FDA recently approved this drug for treatment of sickle cell anemia. It helps in reducing the frequency of pain crises.
- Crizanlizumab (Adakveo). The FDA recently approved this drug for treatment of sickle cell anemia. Given through a vein, it helps reduce the frequency of pain crises. Side effects can include nausea, joint pain, back pain and fever.
- Pain-relieving medications. Your doctor might prescribe narcotics to help relieve pain during sickle cell pain crises.
- Voxelotor (Oxbryta). The Food and Drug Administration (FDA) recently approved this oral drug to improve anemia in people with sickle cell disease. Side effects can include headache, nausea, diarrhea, fatigue, rash and fever.
- Surgical Approaches
- Blood transfusions. In a red blood cell transfusion, red blood cells are removed from a supply of donated blood, then given through a vein to a person with sickle cell anemia. This increases the number of normal red blood cells, which helps reduce symptoms and complications.
Risks include an immune response to the donor blood, which can make it hard to find future donors; infection; and excess iron buildup in your body. Because excess iron can damage your heart, liver and other organs, if you undergo regular transfusions, you might need treatment to reduce iron levels.
- Stem cell transplant. Also known as bone marrow transplant, this procedure involves replacing bone marrow affected by sickle cell anemia with healthy bone marrow from a donor. The procedure usually uses a matched donor, such as a sibling, who doesn't have sickle cell anemia.
Because of the risks associated with a bone marrow transplant, the procedure is recommended only for people, usually children, who have significant symptoms and complications of sickle cell anemia.
The procedure requires a long hospital stay. After the transplant, you'll receive drugs to help prevent rejection of the donated stem cells. Even so, your body might reject the transplant, leading to life-threatening complications.
Preventing infections
Children with sickle cell anemia might receive penicillin between the ages of about 2 months old until at least age 5. Doing so helps prevent infections, such as pneumonia, which can be life-threatening to children with sickle cell anemia.
Adults who have sickle cell anemia may need to take penicillin throughout their lives, if they've had pneumonia or surgery to remove the spleen.
Childhood vaccinations are important for preventing disease in all children. They're even more important for children with sickle cell anemia because their infections can be severe.
See also:
- Mayo Clinic, Sickle Cell Anemia Opens in new window .
- Judy Monroe Peterson, “Sickle Cell Anemia”
