
Sickle Cell Disease
Pathophisiology and Management of Sickle Cell Disease
|
Sickle cell disease (SCD) is an incurable genetic disorder associated with a specific genetic mutation found predominantly in people of specific ethnic background. In addition to Africans and their descendants, Greeks, Italians (especially Sicilians), Eti-Turks, Arabs, southern Iranians, and Asian Indians have a high incidence of this condition. |
By definition, Sickle Cell Disease (SCD) is a group of inherited, autosomal recessive disorders characterized by an abnormal form of hemoglobin in the red blood cell (RBC).
The term sickle derives from the crescent or sickle-shaped red blood cells that are found in different forms in those who have SCD. Because this is a genetic disorder, SCD is usually identified during infancy or early childhood. In some parts of Africa, the incidence is 1 in 40 at birth, with a carrier frequency of 1 in 3. The high frequency of the trait is attributed to the selective advantage it provides against a form of malaria.
SCD results from a mutation in hemoglobin (Hb), in which sickled Hb (HbS) replaces normal adult Hb (HbA) and has a reduced oxygen-carrying ability. As a result, the red blood cells (RBCs) have a decreased lifespan. The sickling of RBCs leads to obstruction of small blood vessels and causes infarcts of the lungs, kidneys, spleen, and bones.
For those who are affected (i.e., those who have SCD), the severity of symptoms varies considerably. Life span is usually foreshortened, with intermittent morbidity.
In general, the sicklelike red blood cells have a reduced survival time, leading to severe anemia and recurrent need for blood transfusions. This serious, chronic hemolytic anemia is incurable and is often fatal by middle age because of renal failure, infection, pulmonary failure, and/or stroke. The abnormal structure of the cells also results in the occlusion of vessels, causing tissue ischemia (lack of oxygen) and severe pain crises.
Most emergency room physicians in urban areas are familiar with patients who require urgent care because of their acute pain from SCD. Although any part of the body and any organ may be affected by blockage of the blood supply, the heart, lungs, kidneys, spleen, pelvic bones, and brain are particularly vulnerable to this damage.
Etiology and Pathophysiology
- Genetic Link
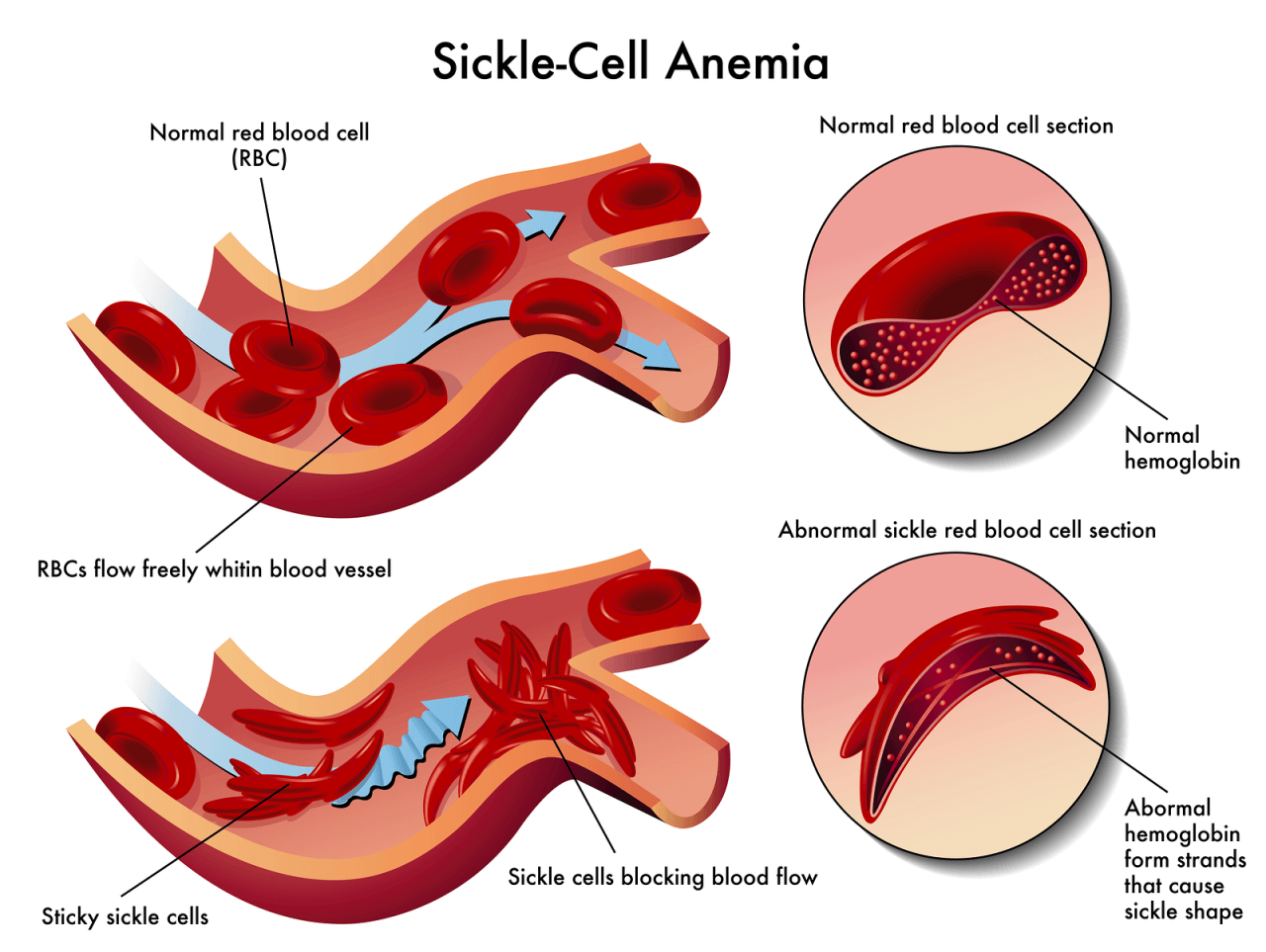
In SCD the abnormal hemoglobin, hemoglobin S (Hgb S), results from the substitution of valine for glutamic acid on the β–globin chain of hemoglobin. Hgb S causes the erythrocyte to stiffen and elongate, taking on a sickle shape in response to low oxygen levels (see Figure X2).
|
| Figure X2 | In sickle cell, the hemoglobin forms long inflexible chains and alters the shape of the red blood cells (RBCs). The sickled RBCs can become stuck in the capillaries and occlude the blood flow. |
SCD consists in several types and includes sickle cell anemia Opens in new window, sickle cell-thalassemia Opens in new window, sickle cell Hgb C disease, and sickle cell trait.
- Sickle cell anemia, the most severe of the SCD syndromes, occurs when a person is homozygous for hemoglobin S (Hgb SS); the person has inherited Hgb S from both parents.
- Sickle cell-thalassemia and sickle cell Hgb C occur when a person inherits Hgb S from one parent and another type of abnormal hemoglobin (such as thalassemia Opens in new window or hemoglobin C) from the other parent. Both of these forms of SCD are less common and less severe than sickle cell anemia.
- Sickle cell trait occurs when a person is heterozygous for hemoglobin S (Hgb AS); the person has inherited hemoglobin S from one parent and normal hemoglobin (Hgb A) from the other parent. Sickle cell trait is typically a mild to asymptomatic condition.
- Sickling Episodes
The major pathophysiologic event of SCD is the sickling of RBCs (see Figure X2). Sickling episodes are most commonly triggered by low oxygen tension in the blood. In patients with SCD, conditions that lead to hypoxia, deoxygenated RBCs, and dehydration can result in acute and sudden episodes of sickle cell crisis.
Sickle cell crisis is a severe, painful, acute exacerbation of RBC sickling causing a vaso-occlusive crisis. As blood flow is impaired by sickled cells, vasospasm occurs, further restricting blood flow.
Severe capillary hypoxia causes changes in membrane permeability, leading to plasma loss, hemoconcentration, thrombi, and further circulatory stagnation. Sickled RBCs become rigid and take on an elongated, crescent shape (see Figure X2).
As a result, sickled cells cannot easily pass through capillaries or other small vessels and can cause vascular occlusion, leading to acute or chronic tissue injury. Tissue ischemia, infarction, and necrosis eventually occur from lack of oxygen.
Shock is a possible life-threatening consequence of sickle cell crisis because of severe oxygen depletion of the tissues and a reduction of the circulating fluid volume. The most common type of sickle cell crisis results from vaso-occlusion, in which tissues become hypoxic, leading to tissue death and pain. Sickle cell crisis can begin suddenly and persist for days to weeks.
Sickle cell crisis can be fatal depending on the part of the body affected and the amount of occlusion. Commonly affected parts of the body include the chest, abdomen, and extremities Opens in new window. Common precipitating factors include viral or bacterial infections, activities in high altitudes, emotional or physical stress, surgery, vomiting, diarrhea, and diaphoresis (Smeltzer & Bare, 2008).
Other events that can trigger or sustain a sickling episode include dehydration, increased hydrogen ion concentration (acidosis), increased plasma osmolality, decreased plasma volume, and low body temperature.
The resulting hemostasis promotes a self-perpetuating cycle of local hypoxia, deoxygenation of more erythrocytes, and more sickling. Circulating sickled cells are hemolyzed by the spleen, leading to anemia Opens in new window. Initially the sickling of cells is reversible with reoxygenation, but it eventually becomes irreversible because of cell membrane damage from recurrent sickling. Thus vaso-occlusive phenomena and hemolysis are the clinical hallmarks of SCD.
Pregnant patients with SCD can experience complications such as pyelonephritis, pregnancy-induced hypertension, urinary tract infections, congestive heart failure, and life-threatening cardiopathology (James et al., 2006). Women with SCD considering pregnancy, a contributor to sickle cell crisis, should seek counseling to understand health risks and the possibility of fetal loss.
The frequency, extent, and severity of sickling episodes are highly variable and unpredictable, but largely depend on the percentage of Hgb S present. Individuals with sickle cell anemia have the most severe form because the erythrocytes contain a high percentage of Hgb S.
Clinical Manifestations
The effects of SCD vary greatly from person to person, the severity of which may be due to genetic polymorphisms. Many people with sickle cell anemia are in reasonably good health the majority of the time. However, they may have chronic health problems and pain because of organ tissue hypoxia and damage (e.g., involving the kidneys or liver).
The typical patient is anemic but asymptomatic except during sickling episodes. Because most individuals with sickle cell anemia have dark skin, pallor is more readily detected by examining the mucous membranes. The skin may have a grayish cast. Because of the hemolysis, jaundice is common and patients are prone to gallstones (cholelithiasis).
The primary symptom associated with sickling is pain. The pain severeity can range from trivial to excruciating. During sickle cell crisis the pain is severe because of ischemia of tissue. The episodes can affect any area of the body or several sites simultaneously, with the back, chest, extremities, and abdomen being most commonly affected. Pain episodes are often accompanied by clinical manifestations such as fever, swelling, tenderness, tachypnea, hypertension, nausea, and vomiting.
Complications
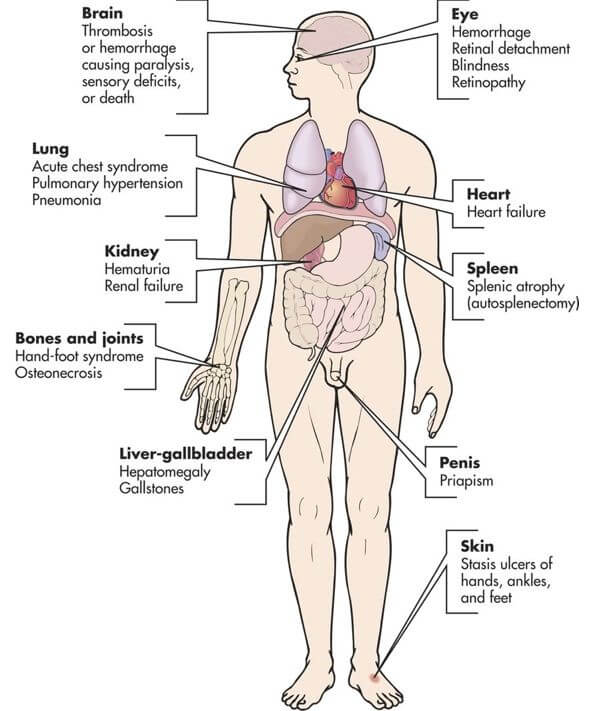
With repeated episodes of sickling, there is gradual involvement of all body systems, especially the spleen, lungs, kidneys, and brain. Organs that have a high need for oxygen are most often affected and form the basis for many of the complications of SCD (see Figure X3).
|
| Figure X3 | Clinical manifestations and complications of sickle cell disease |
Infection is a major cause of morbidity and mortality in patients with SCD. One reason for this is the failure of the spleen to phagocytize foreign substances as it becomes infarcted and dysfunctional (usually by 2 to 4 years of age) from the sickled RBCs. The spleen becomes small because of repeated scarring, a phenomenon termed autosplenectomy.
Pneumonia is the most common infection and often is of pneumococcal origin. Infections can be so severe that they cause an aplastic and hemolytic crisis and gallstones. Aplastic crisis can be so severe that it causes a temporary shutdown of RBC production in the bone marrow.
Acute chest syndrome is a term used to describe acute pulmonary complications that include pneumonia, tissue infarction, and fat embolism. It is characterized by fever, chest pain, cough, pulmonary infiltrates, and dyspnea.
Pulmonary infarctions may cause pulmonary hypertension, MI, HF, and ultimately cor pulmonale. The heart may become ischemic and enlarged, leading to HF. Retinal vessel obstruction may result in hemorrhage, scarring, retinal detachment, and blindness. The kidneys may be injured from the increased blood viscosity and the lack of oxygen, and renal failure may occur.
Pulmonary embolism or stroke can result from thrombosis and infarction of cerebral blood vessels. Bone changes may include osteoporosis and osteosclerosis after infarction. Chronic leg ulcers can result from the hypoxia and are especially prevalent around the ankles. Priapsim (persistent penile erection) may occur if penile veins become occluded.
Diagnostic Studies
A peripheral blood smear may reveal sickled cells and abnormal reticulocytes. The presence of Hgb S can be diagnosed by the sickling test, which uses RBCs (in vitro) and exposes them to a deoxygenation agent.
As a result of the accelerated RBC breakdown, the patient has characteristic clinical findings of hemolysis (jaundice, elevated serum bilirubin levels) and abnormal laboratory test results. Hemoglobin electrophoresis may be done to determine the amout of hemoglobin S.
Skeletal x-rays demonstrate bone and joint deformities and flattening. Magnetic resonance imaging (MRI) may be used to diagnose a stroke caused by blocked cerebral vessels from sickled cells.
Doppler studies may be ued to assess for deep vein thromboses. Other tests may be indicated, such as a chest x-ray, to diagnose infection or organ malfunction.
Nursing and Collaborative Management of Sickle Cell Disease
Collaborative care for a patient with SCD is directed toward
- alleviating the symptoms from the complications of the disease;
- minimizing end-organ damage; and
- promptly treating serious sequelae, such as acute chest syndrome, that can lead to immediate death.
Teach patients with SCD to avoid high altitudes, maintain adequate fluid intake, and treat infections promptly. Pneumovax, Haemophilus influenza, influenza, and hepatitis immunizations should be administered. Chronic leg ulcers may be treated with rest, antibiotics, warm saline soaks, mechanical or enzyme debridement, and grafting if necessary.
Priapism is managed with pain medication, fluids, and nifedipine (Procardia). If it does not resolve within a few hours, a urologist can be called to inject the corpus cavernosum with a dilute solution of epinephrine to preserve penile function.
Sickle cell crises may require hospitalization. Oxygen may be administered to treat hypoxia and control sickling. Because respiratory failure is the most common cause of death, vigilantly assess for any changes in respiratory status.
Rest may be instituted to reduce metabolic requirements, and deep vein thrombosis prophylaxis (using anticoagulants) should be prescribed. Fluids and electrolytes are administered to reduce blood viscosity and maintain renal function.
Transfusion therapy is indicated when an aplastic crisis occurs. Aggressive total RBC exchange transfusion programs may be implemented for patients who have frequent crises or serious complications such as acute chest syndrome. These patients, like those with thalassemia major, may require iron chelation therapy to reduce transfusion-produced iron overload.
Undertreatment of sickle cell pain is a major problem. Lack of understanding can lead health care professionals to underestimate how much pain these patients suffer. Patients, because of their prior opioid treatment, may be tolerant, and thus large doses may be needed to reduce the pain to a tolerable level.
During an acute crisis, optimal pain control usually includes large doses of continuous (rather than as-needed [PRN]) opioid analgeics along with breakthrough analgesia, often in the form of patient-controlled analgesia (PCA). Morphine and hydromorphone are the drugs of choice. Meperidine (Demerol) is contraindicated because high doses can lead to the accumulation of a toxic metabolite, normeperidine, which can cause seizures Opens in new window.
Because patients may be experiencing different types and sites of pain, a multimodal and interdisciplinary approach should be used that incorporates emotional aspects of pain. Adjunctive measures, such as nonsteroidal anti-inflammatory agents, antineuropathic pain medications (e.g., tricyclic antidepressants, antiseizure medications), local anesthetics, or nerve blocks may be used.
Although pain is the most common symptom of patients with SCD seeking medical care, infection is a frequent complication and must be treated. Patients with acute chest syndrome are treated with broad-spectrum antibiotics, O2 therapy, fluid therapy, and possibly exchange transfusion. Blood transfusions have little if any role in the treatment between crises, since patients develop antibodies to RBCs and iron overload. However, because chronic hemolysis results in increased use of folic acid stores, routine oral folic acid should be taken.
Although many antisickling agents have tried, hydroxyurea (Hydrea), which is a chemotherapy drug, is the only one that has been shown to be clinically beneficial. This drug increases the production of hemoglobin F (fetal hemoglobin), decreases the reactive neutrophil count, increases RBC volume and hydration, and alters the adhesion of sickle RBCs to the endothelium. The increase in Hgb F is accompanied by a reduction in hemolysis, an increase in hemoglobin concentration, and a decrease in sickled cells and painful crises.
Hematopoietic stem cell transplantation (HSCT) is the only available treatment that can cure some patients with SCD. The selection of appropriate recipients, scarcity of appropriate donors, risk, and cost-effectiveness limit the use of HSCT for SCD.
Patient teaching and support are important in the long-term care of the patient with SCD. The patient and caregiver must understand the basis of the disease and the reasons for supportive care and ongoing pertinent screening tailored to SCD manifestations, such as eye examinations.
Caregivers should teach the patient ways to avoid crises, including taking steps to avoid dehydration and hypoxia, such as avoiding high altitudes and seeking medical attention quickly to counteract problems such as upper respiratory tract infections.
Teaching about pain control is also needed because the pain during a crisis may be severe and often requires considerable analgesia. Minor pain episodes that are not associated with infection or other symptoms warranting medical attention can sometimes be managed at home.
Adequate rest and sleep and stress reduction techniques may be beneficial. Women may need counseling about sterilization options and alternative contraceptive measures to decrease the risk for pregnancy.
Recurrent episodes of severe acute pain and unrelenting chronic pain can be disabling and depressing. The quality of life of patients with SCD can be profoundly affected. Occupational and physical therapies can help the patient achieve optimum physical functioning and independence. Cognitive behavioral therapy may help patients with SCD cope with anxiety and depression. Support groups may also be helpful.
See also:
- James E. Bowman and Robert F. Murray, Genetic Variation and Disorders in Peoples of African Origin (Baltimore, MD: Johns Hopkins University Press, 1990), p. 192.
- Mabel Koshy, “Sickle Cell Disease and Pregnancy,” Blood Review 9, 3 (1995): 157-64, and Bowman and Murray, p. 198.
- J. B. S. Haldane, “The Rate of Mutation of Human Genes,” Proceedings of the Seventh International Congress of Genetics Heredity 35 (1949, suppl.): 267, and A. C. Allison, “Protection Afforded by Sickle-Cell Trait against Subtertian Malarial Infection,” British Medical Journal 1 (1954): 290-94.
- Peter T. Rowley, S. Loader, C. J. Sutera, M. Walden, and A. Kozyra, “Prenatal Screening for Hemoglobinopathies I: A Prospective Regional Trial,” American Journal of Human Genetics 48 (1991): 439-46.
- M. A. Durosinmi, A. I. Odebiyi, I. A. Adediran, N. O. Akinola, D. E. Adegorioye, and M. A. Okunade, “Acceptability of Prenatal Diagnosis of Sickle Cell Anaemia (SCA) by Female Patients and Parents of SCA Patients in Nigeria,” Social Science and Medicine 41, 3 (1995): 433-36.
- Shirley A. Hill, “Motherhood and the Obfuscation of Medical Knowledge: The Case of Sickle Cell Disease,” Gender and Society 8, 1 (1994): 29-47.
- James E. Bowman, “Genetic Screening and Public Policy,” Phylon 38 (1977): 123.
- Daniel J. Kevles, In the Name of Eugenics: Genetics and the Uses of Human Heredity (New York: Alfred A. Knopf, 1985), p. 278.
- James E. Bowman and Eugene Goldwasser, Sickle Cell Fundamentals (Chicago: University of Chicago Press, 1975), p. 9.
