
Xeroderma Pigmentosum
Introduction and Clinical Features
Xeroderma pigmentosum (XP), an autosomal recessive disease with defective nucleotide excision repair (NER), is a rare sun-sensitive skin cancer syndrome, resulting in serious disfigurement unless stringent protection from sun exposure was practiced.
Xeroderma pigmentosum (XP) is characterized by easily recognizable clinical hallmarks (Table X-1).
| Clinical Hallmarks of Xeroderma Pigmentosum |
|---|
|
These manifestations are due to cellular hypersensitivity to ultraviolet (UV) radiation resulting from a defect in DNA repair. Two types of NER exist: global genome (GG-NER) and transcription coupled (TC-NER).
Eight complementation groups, XPA-XPG, corresponding to defects in the corresponding gene products of XPA-XPG genes and XP-variant, have been described.
These entities occur with different frequencies (e.g. XPA is relatively common, whereas XPE is fairly rare) and they differ with respect to disease severity (e.g., XPG is severe, whereas XPF is mild) and involvement of skin, central nervous system and opthalmological manifestations (Table X-2).
| Table X-2 | Characteristics of Xeroderma Pigmentosum Complementation Groups | |||||
|---|---|---|---|---|---|
| Comple- mentation Group | Frequency (%) | Skin Cancer | Neurological Involvement | Ophthalmological Involvement | Gene Defect |
| A | 30 | + | +++ | + | XPA |
| B | 0.5 | + | + | + | XPB/ERCC3 |
| C | 27 | + | – | + | XPC |
| D | 15 | + | +++ | + | XPD/ERCC2 |
| E | 1 | + | – | + | XPD/ERCC2 |
| F | 2 | + | – | + | DDB2/XPE/p48 |
| G | 1 | + | + | + | XPG/ERCC5 |
| Variant | 23.5 | + | – | + | XPV/hRAD30 |
Cockayne syndrome rarely occurs together with XPB, XPD and XPG.
In addition to the DNA repair defects, UV radiation also exerts pronounced immunosuppressive effects that are likely to be involved in the pathogenesis of XP.
Although typical symptoms of immune deficiency, such as multiple infections, are not usually observed in patients with XP, prominent depletion of Langerhans cells, induced by UV radiation, has been described in XP patients.
Various other defects in cell-mediated immunity such as impaired cutaneous responses to recall antigens, impaired lymphocyte proliferative responses to mitogens and decreased production of interferon as well as reduced natural killer cell activity have been detected in XP patients.
Epidemiology
The frequency of XP in the United States is about 1 case/250.000 inhabitants. Not uncommonly, parental consanguinity and familiarity are present in patients with XP.
XPC is the most common group in the United States, constituting almost 1/3 of XP patients. The unscheduled DNA synthesis is usually between 15-30% of normal. Symptoms for neurological disorders are rare in XP-C.
XPD is the second most common type of XP in the United States and accounts for the majority of US patients with symptoms for neurological disorder being present in about half of all those patients, while the cutaneous and immunologic presentations are quite heterogeneous.
Internationally, the incidence of XP is about the same in Europe, whereas it is higher (1:40.000) in Japan, where XPA is the most common group.
In Europe XPA and XPC are the two most prevalent forms of XP. There is no gender preference. As an autosomal recessive disorder Opens in new window, there is usually no positive family history as the heterozygous parents are clinically healthy.
Dermatological Manifestations
In general, skin problems proceed neurological and ophthalmological symptoms. Several key cutaneous features are usually found. While babies are normal at birth, in the first years of life, diffuse erythema, scaling and pronounced freckle-like pigmentation develop (Figure X-1).
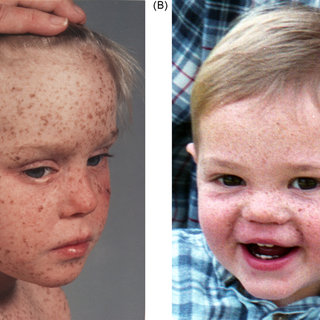
In accordance with the increased light sensitivity, changes are seen over light-exposed areas, in particular face, head and neck and in severe cases they subsequently appear in the lower legs and even the trunk.
One needs to become alert, when babies present with severe solar dermatitis/sunburn, often associated with constant crying, for which no other explanation can be found.
The sunburn will usually persists for extended periods of time, not uncommonly, for several weeks and may include blister formation upon minimal sun exposure.
Once the erythema Opens in new window has resolved, multiple freckles on sun-exposed skin areas cause mottled pigmentation, telangiectasias and actinic damage. XP is one of the few diseases that can cause poikiloderma at an early age.
Poikiloderma Opens in new window is characterized by erythema, hyper- and hypopigmentation as well as scarring and telangiectasias. As the skin suffers actinin damage, the surface become atrophic and dry, which has led to the term xeroderma (dry skin) for this condition.
The incidence of tumors is about 1000-fold increased as compared with the normal population.
The process of malignant transformation in XP has been estimated to be around 8 years as compared with 60 years for a representative control cohort. The mean patient age of developing skin cancer is 8 years in XP patients and for the onset of actinin damage around 1-2 years of age.
Characteristically, children or adolescents develop large areas with field cancerization including multiple actinin keratoses, in situ squamous cell cancer and malignant skin tumors.
Upon the malignant tumors, the UV light-induced cancers like squamous cell cancer, basal cell cancer and lentigo maligna melanomas are predominating.
Especially for melanoma, the role of UVB is widely accepted. Interestingly, the total UV-dose plays an important role in lentigo malignant melanoma in contrast to other malignant melanomas.
The important role of UV light has also been documented. In addition, UV radiation has immunosuppressive effects, which are important for the control of premalignant and malignant cells.
Despite the fact that tumor cells express tumor-associated antigens, that are recognized by specific T-cells, tumor progression occurs. The danger associated with the development of malignant melanoma and squamous cell carcinoma is their early metastases.
Other Cancers in Xeroderma Pigmentosum
Besides the above mentioned cancers, keratoacanthomas and sarcomas including fibrosarcomas and angiosarcomas have been described. The incidence of tumors of the oral mucosa (inner lips, tongue, gingival) and other organs (brain and lung cancer and leukemia) is also increased. Particularly, the occurrence of leukemias has been reported in XP. Other oral manifestations include caries of primary dentation.
Neurological Manifestations
Neurological abnormalities have been described in 18% of 830 patients in the largest study available to date with mental retardation being present in 80% of these individuals. The median intelligence quotient score was 45 with a range of 15-81.
The second most frequent neurological abnormality was spasticity or ataxia (30% of subjects with neurological involvement), followed by microcephaly (24%).
Patients with neurological symptoms can be classified on the basis of age at the onset of neurological symptoms: juvenile (before age 20 years) and adult (after 20 years).
As a general rule, the presence of neurological abnormalities correlates with the degree of NER defects; patient with the greatest impairment of DNA repair (i.e., complementation groups XPA and XPD; Table X-2) are more prone to develop neural degeneration.
As many as 50% of patients with XPD manifest neural deterioration, while on the contrary, neurological involvement is rare in patients with XPC, which is the most common complemention group in the United States.
In general, Central Nervous System (CNS) involvement is due to premature neuronal death due to damage resulting from DNA damage from reactive oxygen species or other free radicals.
Neurodegeneration probably results from accumulating mutations due to cells’ inability to repair DNA damage in response to oxidative stress. Neurological symptoms are progressive and irreversible. Therefore, many patients develop severe disability in the sense that they become bed-ridden and incontinent with infections, sepsis and aspiration pneumonia as potential complications.
Neurological problems include reduced intelligence (80%) with a median IQ test result of 45, abnormal motor activity (30%) with hyporeflexia, areflexia, ataxia, chorea, spasticity as well as peripheral neuropathy and impaired hearing.
For example, in 32 Japanese patients with XPA, the most profound DNA repair defect, mental retardation, microcephaly, nystagmus, dysarthria, ataxia and short stature wree described as the most prominent neurological manifestations.
Half of the patients with neurological abnormalities have skin cancer. Not uncommonly, sexual development is delayed or absent in 12% in XP patients with neurological disorders.
The incidence of central nervous system tumors is about 10-fold higher than in normal individuals. The neurological tumors include astrocytoma, meduloblastoma, glioblastoma and malignant schwannoma.
The de Sanctis-Cacchione-syndrome refers to cutaneous XP, neurological abnormalities, hypogonadism and dwarfism. These children are characterized by small stature, spasticity and debility. Sometimes, additional symptoms such as segmental demyelinisation, microcephaly, sensory deafness and epilepsy may also occur.
Ophthalmological Manifestations
About 40% of XP patients suffer from ophthalmological problems. The photosensitivity in XP is variable, but usually more pronounced in the range of 290-320 nm (UVB range). However, the minimal erythema dose is generally lower than normal at other wavelengths.
Among the ocular tissues, the lids, conjunctiva and cornea receive substantial amounts of UV light and are therefore important target sites in XP patients. Important ocular problems include photophobia, conjunctivitis (with conjunctival infection?) and corneal abnormalities.
Blepharitis, ectropion, symblepharon, loss of eyelashes, atrophy and scarring represent some less common ophthalmological features in XP patients.
Corneal abnormalities include corneal clouding, vascularization and corneal ulcers causing impaired vision in about 15% are some less common ophthalmological features in XP patients.
The risk of ocular neoplasias such as basal cell cancer and squamous cell cancer is increased about 200-fold. These cancers occur in up to 15% of patients and are most often localized on the cornea and conjunctiva, while malignant melanoma occurred in about 5%. In addition, fibrovascular pannus of the cornea and epitheliomas of the lids and conjunctivae may occur.
Differential Diagnoses
Differential diagnoses of XP include several other rare syndromes, which can be ruled out by serologic, genetic and metabolic tests. Some overlaps may be seen with Cockayne syndrome (CS) and trichothiodystrophy (TTS).
Both diseases are of autosomal recessive inheritance and are characterized by increased X-rays (or light) sensitivity, while CS patients present with characteristic neurological symptoms, ataxia, mental retardation and sensory deafness, distinct facial features with large, halonated eyes, prominent nose, progressive cachexia, myopathy, short stature, microcephaly and pigmented degeneration of the retina as well as optic atrophy and cataract.
Recently, some overlaps have been detected between CS and TTD with XP. In CS, there is a defect in TCR, but the global genome repair is functional. CS shows no increased risk of skin cancer.
The characteristic hallmarks of TTD are short and fragile hair, ichthyosis and short stature. Interestingly, the hair shows a deficit of sulfur-rich proteins. The hair has a characteristic tiger pattern under the polarizing microscope. Additional symptoms include reduced intelligence and compromised fertility and certain defects in XP-B and XP-D genes.
Diagnosis of XP
Dermatological manifestations serve to make the diagnosis. The clinical features include the pathological photosensitivity at an early age are the hallmarks of the disease. No consistent routine laboratory abnormalities are present in XP.
Specialized laboratories may analyze the sensitivity of cultured fibroblasts to UV radiation and chromosomal breakage, complementation studies and gene sequencing to identify the specific molecular defect. Ideally, the chromosomal breakage is compared with the patients’ parents cells as they are obligate heterozygotes for XP.
Prenatal diagnosis possible; however, due to the recessive nature of the disease, there may be no suggestion of such disease in the family history. For prenatal testing, unscheduled DNA synthesis serves as the classical method for the diagnosis of XP.
There is no specific histology of XP; usually melanocytic nevi are numerous and increased melanin pigments are present in the basal cell layer together with a chronic inflammatory infiltrate in the upper dermis.
At later stages, the histological picture of poikiloderma may be seen including hyperkeratosis, atrophy, hyperpigmentation and telangiectasias. The dermis usually shows elastotic changes. Likewise, there is no specific test for CNS and eye involvement.
Even in patients with neurological symptoms, the electroencephalographic findings may be normal. Electromyography (EMG) and nerve conduction studies (NCS) are helpful because axonal polyneuropathy is common in XP.
In addition, several metabolic studies are recommended to exclude neurological manifestations mimicking XP and CNS involvement. Therefore, serum and urine amino acids, serum copper, lysosomal enzymes, cholesterol esterification, mucopolysaccharides as well as long-chain fatty acid analyses should be performed besides lactate and pyruvate concentration to rule out other CNS conditions.
Management
As a curative therapy such as gene therapy is currently not available, the overall preventive measure is to keep the patient away from sunlight. This includes shifting the daily activities into the night as much as possible; this has led to the term “moon babies.” In addition, protective clothing, hats and appropriate eye care can serve to minimize UV-induced damage.
Sunscreens should be regularly applied to all exposed surfaces, including the hands and the lower limbs. Preferably, physical and chemical sunscreens are to be used simultaneously and around the entire year and during evening as well as early morning hours.
Patient education about effective sun protection and early recognition of skin cancers is also helpful. Counseling can also be offered by the XP society (www.xps.org). prevention of skin cancer in XP patients has also been achieved to some degree with the use of oral isotretinoin.
Recent attempts to repair DNA damage after UV exposure have been made by topical delivery of DNA repair enzymes to the skin by means of specially engineered liposomes (T4 endonuclease-V). careful dermatologic examinations are mandatory at regular intervals of 3-6 months.
Prognosis
Until today, early diagnosis and consequent avoidance of sunlight as well as regular dermatological screening have helped to increase the life expectancy. The prognosis is significantly impaired as fewer than 40% of patients survive beyond 20 years of age. However, some individuals with milder disease may survive to about 40 years.
Ken Kraemer and colleagues have constructed Kaplan-Meier survival curves from 830 published XP patients, describing a 90% probability of surviving to age 13 and 80% of surviving to age 28 and a 70% probability surviving to 40 years.
Overall, the life expectancy for XP patients was reduced by 30 years. Patients are likely to die from cancer (33%), infections (11%) and various other diseases. Heterozygote carriers have no increased risk to develop skin tumors. Subsequently to the chronic occurrence of the aforementioned skin cancers, some degree of mutilation may develop, which may be severe in certain patients.
- Jimbo T, Ichihashi M, Mishima Y et al. Role of excision repair in UVB-induced depletion and recovery of human epidermal Langerhans cells. Arch Dermatol 1992; 128:61-62.
- Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous ocular and neurologic abnormalities in 830 published cases. Arch Dermatol 1987; 123:241-250.
- Goyal JL, Rao VA, Srinivasan R et al. Oculocutaneous manifestations in xeroderma pigmentosa. Br J Ophthalmol 1994; 78:295-297.
- English JS, Swerdlow AJ. The risk of malignant melanoma, internal malignancy and mortality in xeroderma pigmentosum patients. Br J Dermatol 1987; 117:457-461.
- Kobayashi M, Satoh Y, Irimajiri T et al. Skin tumors of xeroderma pigmentosum (I). J Dermatol 1982; 9:319-322.
- Kraemer KH, Lee MM, Scotto J. Early onset of skin and oral cavity neoplasms in xeroderma pigmentosum. Lancet 1982: 1:56-57.
- van Steeg H, Mullenders LH, Vijg J. Mutagenesis and carcinogenesis in nucleotide excision repair-deficient XPA knock our mice. Mutat Res 2000; 450:167-180.
- Morison WL, Bucana C, Hashem N et al. Impaired immune function in patients with xeroderma pigmentosum. Cancer Res 1985; 45:3929-3931.
- De Silva BD, Nawroz I, Doherty VR. Angiosarcoma of the head and neck associated with xeroderma pigmentosum variant. Br J Dermatol 1999; 141:166-167.
- Hakamada S, Watanabe K, Sobue G et al. Xeroderma pigmentosum: neurological, neurophysiolotical and morphological studies. Eur Neurol 1982; 21:69-76.
- Robbins JH. Xeroderma pigmentosum. Defective DNA repair causes skin cancer and neurodegeneration. JAMA 1988; 260:384-388.
