
CHILD Syndrome
Clinical Definition and Features
The CHILD syndrome is a congenital disorder characterized by congenital hemidysplasia, ichthyosiform erythroderma, and limb defects. CHILD, therefore, is an acronym for Congenital Hemidysplasia with Ichthyosiform nevus and Limb Defects.
Also known as Unilateral CIE, the hallmark of the disorder is the sharp midline demarcation and its largely unilateral cutaneous and skeletal features.
The syndrome was delineated by Happle et al. in 1980 as an X-linked dominant trait characterized by the phenomenon of functional X-chromosome mosaicism Opens in new window.
In the original publication the ‘I’ stood for Ichthyosiform erythroderma Opens in new window. However, characteristic and localized cutaneous pathology of this syndrome represents an entity from ichthyosiform erythrodermas that should be referred to as CHILD nevus.
This X-linked dominant condition occurs almost exclusively in girls and is presumed to be lethal in affected males. The only case in a boy is thought to represent early postzygotic mosaicism.
Skin Features
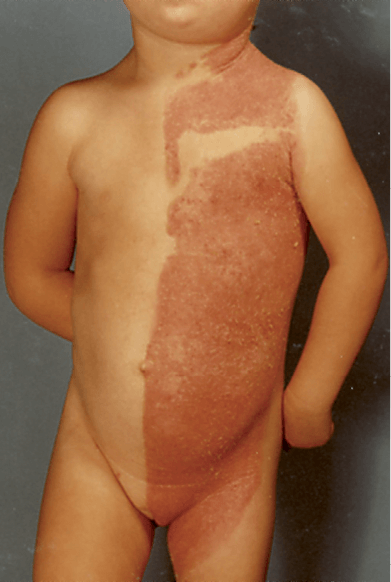
The most striking feature is an inflammatory nevus with a unique lateralization and strict midline demarcation characterized by yellowish, wax-like scales that can be removed without bleeding.
This nevus may sometimes partially resolve later in life, but usually it shows a pronounced affinity for the body folds, a phenomenon to which the term ptychotropism has been applied.
In some cases, the nevus follows the lines of Blaschko Opens in new window, but there is virtually always a far more pronounced involvement of one side of the body.
Ipsilateral hypoplasia of the body may affect all skeletal structures, and shortness or even complete absence of a limb is noted.
Development of visceral organs may be impaired on the affected body side; defects of kidney, lung, heart and brain have been reported.
Unilateral alopecia Opens in new window and severe nail dystrophy Opens in new window with claw-like nails have been observed. The face is typically spared. With increasing age, lesions may improve or even clear spontaneously, but lesions in intertriginous areas (ptychotropism) tend to persist and be most severely affected sites.
Histopathological examination shows a psoriasiform dermatitis Opens in new window with acanthosis Opens in new window, zones of parakeratosis and an exocytosis of neutrophils that may form accumulations reminiscent of Munro abscesses Opens in new window.
Apart from these epidermal changes, the distinctive feature of verruciform xanthoma Opens in new window may be found in the dermis, especially when the biopsy is obtained from a body fold. The widened dermal papillae abound with foamy histiocytes. Ultrastructurally, these cells show intracytoplasmic vacuoles Opens in new window that contain large amounts of so far unindentified lipids.
Skeletal Features
The skeletal manifestations include the phenomenon of epiphyseal stippling Opens in new window, which can only be seen on X-rays performed in early infancy. Shortening or absence of a limb is the most striking manifestation but many patients also show involvement of the spine or the pelvis.
The degree of skeletal involvement may vary considerably and sometimes shortening of a single phalanx may be the only clinical finding. Visceral defects are noted on the side of CHILD nevus and limb malformations. Absence of a kidney, hypoplasia of lung, thyroid or heart, have been resolved.
Involvement of the nervous system includes ipsilateral defects of cranial nerves, pons, medulla, cerebellum, or spinal cord. The electroencephalogram may show ipsilateral abnormalities.
The underlying molecular defect has been elucidated. Point mutations as well as deletions in the gene NSDHL (AD(P)H steroid dehydrogenase-like protein) at Xq28 were demonstrated in more than 20 patients with CHILD syndrome.
This gene most likely encodes a novel type of 3β-hydroxysteroid dehydrogenase, similar to that described in the murine Nsdhl mutant bare patches.
The molecular analysis provides a reliable tool for genetic counseling, since mother-to-daughter transmission has sometimes been reported.
Moreover, it is possible to confirm the diagnosis in patients with minimal or atypical involvement, for instance in an exceptional case with almost symmetricl manifestations and in a family with mild CHILD syndrome in three generations.
Patterned involvement is seen in female carriers of many X-linked genodermotes such as focal dermal hypoplasia, X-linked dominant chondrodysplasia punctata or incontinentia pigmenti Opens in new window.
The concept of functional X-chromosome mosaicism can be applied to these phenotypes. Due to the Lyon effect of X-inactivation, two functionally different cell clones develop at an early stage of embryogenesis leading to a patterned involvement of streaks and patches.
However, the striking lateralization pattern as seen in CHILD syndrome is unique. Probably the NSDHL gene plays an important role in early human embryogenesis and the gene product is likely to be involved in the development of the bilateral body symmetry.
- Happle R, Koch H, Lenz W. The CHILD syndrome: congenital hemidysplasia with ichthyosiform erythroderma and limb defects. Eur J Pediatr. 1980;134:27.
- König A, Happle R, Bornholdt D, et al. Mutations in the NSDHL gene, encoding a 3β-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am J Med Genet. 2000;90:339.
- Bornholdt D, König A, Happle R, et al. Mutational spectrum of NSDHL in CHILD syndrome. J Med Genet. 2005;42:317.
- Bittar M, Happle R, Grzeschik KH, et al. CHILD syndrome in 3 generations: the importance of mild or minimal skin lesions. Arch Dermatol. 2006;142:348-351.
