
Hemophagocytic Lymphohistiocytosis (HLH)
Clinical Definition and Features
Hemophagocytic lymphohistiocytosis (HLH) comprises a heterogeneous group of primary (familial) and secondary (non-famial) disorders characterized by the proliferation of activated macrophages associated with generalized hemophagocytosis. Other applicable terms to describe this syndrome include cytokine disease and macrophage activation syndrome.
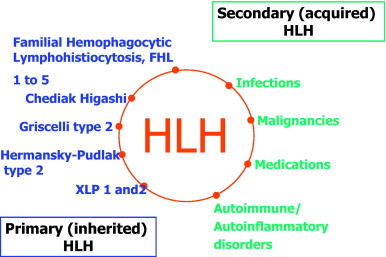
Within the familial group, five types of disorders—all inherited as autosomal recessives Opens in new window—have been identified: familial hemophagocytic lymphohistiocytosis (FHL), Chediak-Higashi syndrome (CHS) Opens in new window, Griscelli syndrome (GS) Opens in new window, X-linked lymphoproliferative syndrome (XLP) Opens in new window or Duncan disease, and lysinuric protein intolerance (LPI) Opens in new window.
The acquired or secondary form of HLH was originally identified as infection associated hemophagocytic syndrome in immune-suppressed patients by Risdall and colleagues (Risdall et al. 1979).
Later, it was recognized that this syndrome was not only seen in immune-suppressed patients, but was associated with a wide variety of disorders in immune-competent as well as immune-suppressed patients. These include non-viral infections, rheumatoid arthritis, and malignant processes such as T-cell malignancies.
EBV infection Opens in new window is a frequent cause of secondary HLH. However, it is important to recognize that EBV infection may trigger the clinical manifestations of FHL and plays a role in the accelerated phase of CHS. Therefore, its demonstration as an etiological agent does not necessarily represent a secondary form of HLH.
Characteristically, familial hemophagocytic lymphohistiocytosis (FHL) presents in infancy and secondary hemophagocytic lymphohistiocytosis develops in older children, although this is not a constant feature.
Clinically, both the primary and secondary forms of HLH present with same clinical features including:
- fever
- hepatosplenomegaly
- PB cytopenias
- coagulopathy
- liver dysfunction
- impaired natural killer (NK) cell activity
- hypertriglyceridemia
- hyperferitinemia
- increased levels of aminotransferases
- lactate dehydrogenase
- soluble interleukin-2 receptor
- interferon gamma
- tumor necrosis factor-α
- interleukin-6 and interleukin-10
- and an increase in activated BM macrophages.
Differential Diagnosis
The clinical and pathological similarities between FHL and secondary HLH make their differentiation very difficult, except in cases with a familial history.
To facilitate the differential diagnosis of HLH syndrome, criteria have been established by the Study Group of the Histiocyte Society, although in the foreseeable future the diagnosis of these disorders will most likely be based on the determination of the genetic defect or primary disorder by new molecular genetic techniques.
The differential diagnosis between FHL and secondary HLH is extremely important. FHL is a fatal disorder unless treatment with allogeneic BMT from a related healthy donor is instituted.
On the other hand, secondary HLH associated with infectious diseases in non-immune-suppressed patients does not require BMT and usually resolves with appropriate treatment of the primary disease with total hematologic recovery.
Secondary HLH associated with malignancies can be subdivided in cases where HLH presents masking a T-cell malignancy or develops during chemotherapy for the malignancy, or is associated with a relapse of a malignancy.
Difficulties in the differential diagnosis between HLH and T-cell malignancies associated with HLH also exist in the area when EBV is the etiological agent of the secondary HLH.
Gene Description
Perforin is a protein expressed by NK cells, cytolytic lymphocytes and macrophages, and it plays a main role in the cytolytic process.
A defect in perforin has been associated with FHL as well as with other inherited syndromes associated with BM hemophagoctytosis such as Griscelli syndrome (GS Opens in new window) and Chediak-Higashi syndrome (CHS) Opens in new window.
A defect in the NK cells is almost always present in children with FHL. A recent report proposes that flow cytomeric studies based on the quantification of NK cells and low expression of perforin by cytotoxic lymphocytes could be used to identify patients and carriers Opens in new window of perforin deficiency.
Moreover, investigators suggest that these studies could provide a diagnostic tool for the differentiation of FHL and EBV-induced HLH.
Within primary FHL two genetic defects have been identified at the molecular level. In one the abnormal gene has been mapped to chromosome 9 (9q21.3-q22) and in the other it has been mapped to chromosome 10 (10q22).
In Chediak-Higashi syndrome Opens in new window the abnormal gene has been mapped to chromosome 1 (1q42.1-42.2). In Griscelli syndrome Opens in new window the abnormal gene has been mapped to chromosome 15 (15q21).
In XLP syndrome Opens in new window the abnormal gene has been mapped to chromosome X (Xq25) and in LPI Opens in new window the defected gene has been mapped to chromosome 14 (14q11.2).
Laboratory Findings
Hemophagocytosis is a common observation in the examination of normal as well as abnormal BM.
Moreover, phagocytosis of hematopoietic cells is not limited to normal or abnormal hematopoietic cells, but may occasionally be observed within neoplastic cells or tumors metastatic to the BM.
It should be noted that although HLH is a generalized disorder that manifests in multiple organs, the best diagnostic tool is examination of the BM. Changes in the PB and BM occur during the course of the disease. In the early stages, PB cytopenia may or may not be present.
BM examination reveals a few histiocytes containing mature or immature hematopoietic cells in their cytoplasm, but in general terms hematopoiesis is preserved.
In later stages, PB bicytopenias or pancytopenia develops. BM examination at this time shows a large number of histiocytes and a decrease in all hematopoietic elements except in some cases, where megakaryocytes may be sparse.
See also:
- Adams ND, Carrera GF, Johnson RP, Latoraraca R, Lemann J, Jr (1982) Calcium-oxalate-crystal-induced bone disease. Am J Kidney Dis 1:294-299.
- Anderson RA, Rao N, Byrum RS, Rothschild CB, Bowden DW, Hayworth R, Pettenati M (1993) In situ localization of the genetic locus encoding the lysosomal acid lipase/cholesteryl esterase (LIPA) deficient in Wolman disease to chromosome 10q23.2-q23.3. Genomics 15:245-247.
- Arico M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, Martinetti M, Rusca MP (1996) Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia 10:197-203.
- Arceci RJ (1999) The histiocytoses: the fall of the Tower of Babel. Eur J Cancer 35:747-767; discussion 767-769.
- Arico M, Danesino C, Pende D, Moretta L (2001) Pathogenesis of haemophagocytic lymphohistiocytosis. Br J Haematol 114:761-769.
- Baetz K, Isaaz S, Griffiths GM (1995) Loss of cytotoxic T lymphocyte function in Chediak-Higashi syndrome arises from a secretory defect that prevents lytic granule exocytosis. J Immunol 154:6122-6131.
