
Osteogenesis Imperfecta
Clinical Features of Type II Osteogenesis Imperfecta
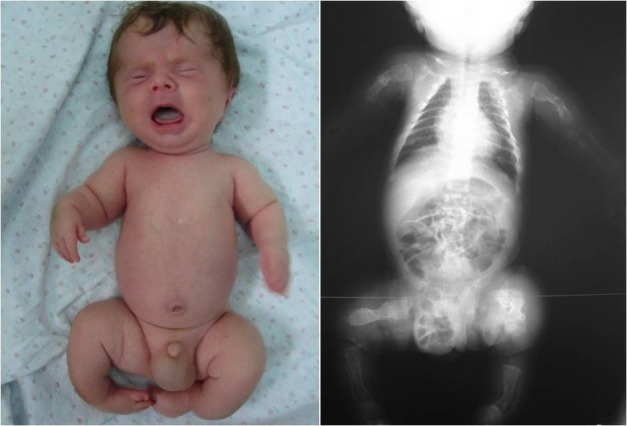 Figure X-1, Congenita A type of osteogenesis imperfecta in a 2-month-old child | Photo courtesy of ResearchGate Opens in new window
Figure X-1, Congenita A type of osteogenesis imperfecta in a 2-month-old child | Photo courtesy of ResearchGate Opens in new window
Type II osteogenesis imperfecta is the most severe form of osteogenesis imperfecta Opens in new window. This form is characterized by extreme bone fragility and frequent fractures, usually resulting in prenatal fatality.
Infants with type II OI experience intrauterine fractures and intracranial hemorrhage following vaginal delivery, and succumb shortly after birth to pneumonia or respiratory insufficiency secondary to decreased thoracic size.
Clinically, in utero fractures are present in 100% of cases. Many are stillborn, and 90% die before four weeks of age. Blue sclera may be present. Hearing loss is not common to type II OI. Dentinogenesis imperfecta Opens in new window may be present along with small nose, micrognathia and short trunk.
Clinical Manifestations
- General features
This clinically and biochemically heterogeneous group of osteogenesis imperfecta syndrome is characterized by extreme bone fragility consistently leading to intrauterine or early infant death.
Sillence et al subclassified this form into three subtypes (groups A, B, and C) distinguished from one another on the basis of radiographic features. Subtype IIA is the most common subtype, IIC the most frequent.
The distinction between the three subtypes is not always clear, as the radiographic findings represent a continuum. In general, the better the bone morphology and mineralization at birth, the longer the survival of the newborns.
- At birth, newborns in group A are small for gestational age and usually premature.
- Twenty percent are stillborn and the remainder die within hours or days of birth; 90% are no longer living by 4 weeks of age.
- Breech delivery occurs in 15%.
- General connective tissue fragility is present and the head or a limb may be torn off during delivery.
Type IIA distinctive features
- Infants in group B have a mean gestational age of 37.6 weeks;
- mean survival is about 14 hours.
Type IIB distinctive features
Length of gestation and survival of individuals in group C have not been described in detail because few examples have been reported; however, two cases reported by Thompson et al were stillborn at 28 and 30 weeks, respectively.
Since most, if not all, cases of osteogenesis imperfecta type II have been ascertained through early death, the true average length of survival is unknown.
Although instances of familial recurrence and parental consanguinity Opens in new window have been observed in group A, the majority are sporadic examples. It was originally thought that most cases have autosomal recessive inheritance Opens in new window.
However, from a study of 30 cases, Young et al concluded that most cases of osteogenesis imperfecta type IIA were the result of new dominant mutations; no sib pairs were ascertained, 25 were born to noncansanguineous couples, and increased paternal age effect was observed. This was later also confirmed by molecular studies.
The sibling recurrence in type IIA can be explained by germline and somatic mosaicism in parents, which has been confirmed by molecular studies.
Thompson et al calculated an empirical recurrence risk of 7.7% for type IIB; no increase in parental age over the general population was found. It seems probable that a proportion of cases arises from de novo autosomal dominant mutations Opens in new window, and another part arises from autosomal recessive inheritance Opens in new window.
Type IIC may follow autosomal recessive inheritance, although too few cases are available for analysis. The empiric overall recurrence risk of type II, irrespective of the subtype, is approximately 6%.
- Facies
The face and cranium are molded and soft and the cranium often appears disproportionately large for the face. Commonly, mild micrognathia and a small narrow nose are noted.
- Ophthalmologic findings
Deep blue-gray sclera are present in virtually all affected infants. On examination of the cornea of an infant who died at 17 days of age with presumed osteogenesis imperfecta type II, collagen fiber diameter was reduced, normal cross-striations were not seen, and collagen fibers were more densely packed than normal.
- Cardiovascular system
Information about the cardiovascular system in osteogenesis imperfecta type II has been derived from autopsy studies.
Abnormalities include:
- thickening of the valve leaflets,
- myxoid degeneration of valves,
- calcification of pulmonary, cerebral, or pulmonary artery,
- thickening of the media and adventitia in small and medium-sized pulmonary arteries, and
- atherosclerotic changes in the aorta.
Microscopc calcification throughout the aorta and endocardium has also been noted.
- Skeletal manifestations
In type IIA, there is marked reduction in ossification of the facial bones and cranial vault, and numerous wormian bones are present.
The chest is small. Ribs are slightly short, thick, and continuously beaded. The humeri and femora are crumpled (accordion-shaped), short, and broad.
Thighs are held in abduction at right angle to the body. Tibias are broad, accordion-shaped, and angulated. Vertebrae are flattened, the ilia are broad and round, and the Ischia and pubic bones are broad and without form.
Few cases of osteogenesis imperfect types IIB and IIC have been studied. In type IIB, the skull exhibits spotty mineralization and the ribs are thin and wavy without beading, but ribs exhibit only occasional beading and are not uniformly thick. Femurs are short, broad, and crumpled, and tibias are thickened and angulated.
In type IIB there are more well-modeled humeri with wide metaphyses, and there is more normal vertebral body height than type IIA.
In type IIC, the face is not remarkably abnormal. The arms are longer than in type IIA, but the legs are similarly bent inward.
Marked underossification of the skull and slender but not so uniformly beaded ribs as in type IIA have been noted. Long bones are slender and shafts are inadequately modeled. Angulation deformities of the shafts of all long bones and multiple fractures are seen. Heights of vertebral bodies are near normal.
- Oral abnormalities
Dental abnormalities have been reported. Dean and Hiramato noted argyrophilic fiber-like structures in the dentin, absence of predentin, irregular pulpal-dentin junction, paucity of argrophilic granules in the odontoblast cytoplasm, abundance of argyrophilic fibers in the coronal pulp, and dilated capillaries in the coronal pulp.
No abnormalities were found in the enamel organ or in the morphology of developing teeth.
Calonius et al reported the dental findings in an infant who died 8 days after birth: the dentin was thinner than normal, interglobular dentin extended to the cemento-enamel junction, dentin tubules in the circumpulpal dentin were few and wide, and degenerated osteoblasts were found entrapped in the dentin. Mantle dentin was normal and irregular at the enamel-dentin interface; the dental papilla and pulp were normal.
Similar findings have been reported elsewhere. Haebara et al, however, reported normal teeth in a newborn with presumed osteogenesis imperfecta type II who died shortly after birth, suggesting heterogeneity.
Levin et al also reported normal dentition in an infant with a lethal osteogenesis imperfecta syndrome of unknown type who survived for 10 months.
Although other studies have reported dental abnormalities in patients with lethal osteogenesis imperfecta, phenotypes have not been sufficiently characterized; thus, it cannot be determined whether they had osteogenesis imperfecta type II or another lethal osteogenesis imperfecta syndrome.
See also:
- Clinical Features of Type I Osteogenesis ImperfectaOpens in new window
- Clinical Features of Type III Osteogenesis ImperfectaOpens in new window
- Clinical Features of Type IV Osteogenesis ImperfectaOpens in new window
- Osteogenesis Imperfecta (Molecular Pathogenesis of the syndrome)Opens in new window
- Therapeutics for Patients with Osteogenesis ImperfectaOpens in new window
- Bauer KH: Uber Osteogenesis imperfecta. Zugleich ein Beitrag zur Frage einer allgemeinen Erkrankung santlicher Stutzgewebe. Dtsch Z Chir 154:166-213, 1920.
- Haebara H et al: An autopsy case of osteogenesis imperfecta congenital—histochemical and electron microscopical studies. Acta Pathol Jpn 19:377-394, 1969.
- Becks H: Histologic study of tooth structure in osteogenesis imperfecta. Dent Cosmos 73:437-454, 1931.
- Bixler D et al: Dentinogenesis imperfecta: Genetic variations in a six-generation family. J Dent Res 49:1196-1199, 1955.
- Calonius PEB et al: Tooth germ changes in osteogenesis imperfecta. A case report. 63:220-226, 1967.
- Chan CC et: Ocular findings in osteogenesis imperfecta congenital. Arch Ophthalmol 100:1459-1463, 1982.
- Byers PH et al: Perinatal lethal osteogenesis imperfecta: Translation of mutation to phenotype. J Med Genet 28:433-442, 1991.
- Sillence DO et al: Osteogenesis imperfecta type II. Delineation of the phenotype with reference to genetic heterogeneity. Am J Med Genet 17:407-423, 1984.
- Kuivaniemi H et al: A 19 base pair deletion in the pro alpha2(1) gene of type I procollagen in a proband with atypical osteogenesis imperfecta and in his asymptomatic mother. J Biol Chem 263:11407-11413, 1988.
- Lund AM et al:Gly802Asp substitution in the pro alpha2(I) collagen chain in a family with recurrent osteogenesis imperfecta due to paternal mosaicism. EUr J Hum Genet 4:9-45, 1996.
- Cohen-Solal L, et al: Dominant mutations in familial lethal and severe osteogenesis imperfecta. Hum Genet 87:297, 1991.
- Dean DH, Hiramato RN: Osteogenesis imperfecta congenital: Dental features of a rare disease. J Oral Med 39:119-121, 1984.
- Cole WG, Dalgleish R: Perinatal lethal osteogenesis imperfecta. J Med Genet 32:284-289, 1995.
- Young ID et al: Osteogenesis imperfecta type IIA: Evidence for dominant inheritance. J Med Genet 24:386-389, 1987.
- Thompson EM et al: Recurrence risks and prognosis in severe sporadic osteogenesis imperfecta. J Med Genet 24:390-405, 1987.
- Levin LS et al: Osteogenesis imperfecta lethal in infancy: Case report and scanning electron microscopic studies of the deciduous teeth. Am J Med Genet 12:359-368, 1982.
