
Sotos Syndrome
Clinical Definition and Features
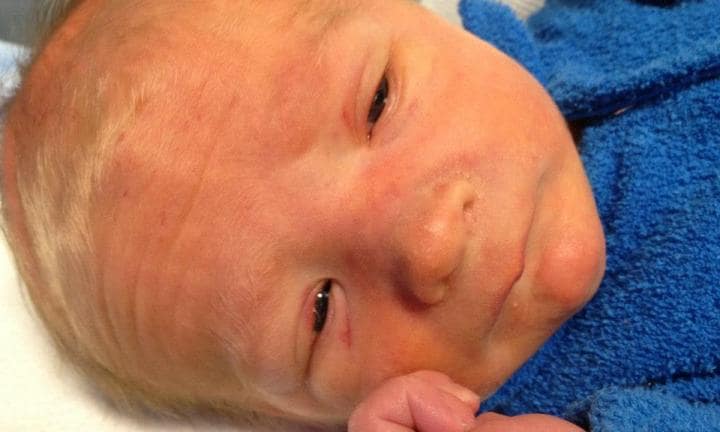 Figure X-1 | Photo courtesy of Kidspot Opens in new window
Figure X-1 | Photo courtesy of Kidspot Opens in new window
Sotos syndrome (also called cerebral gigantism), first described by Sotos et al in 1965, is an overgrowth syndrome characterized by increased birth length, increased birth weight, excessive growth during the first four years of life, advanced bone age, and distinctive facial features including flat face, hyperteloric eyes and prominent jaw. Also, the cranium has a circumference above two standard deviations and is dolichocephalic.
The Sotos syndrome, often occurring sporadically, is relatively common among overgrowth syndromes and appears to occur ten times more frequently than Weaver syndrome Opens in new window.
Identical twins have been discordant for the syndrome; and several reported families have been consistent with autosomal dominant inheritance Opens in new window.
Schrander-Stumpel found a balanced translocation: t(3;6)(p21;p21). Breaking point mapping has been carried out on this patient. This is in part supported by the observation of Cole et al that there is loss of heterozygosity at 3p21 in small cell lung carcinoma that has been found in Sotos syndrome.
The syndrome is associated with developmental delay and children tend to be clumsy. Main clinical findings are discussed below.
Clinical Manifestations
- Growth and Skeletal Findings
Overgrowth is commonly manifested in the newborn, birth weight averaging 4200 g in males and 4000 g in females.
- Birth length, birth weight, and head circumference are, respectively, 3.2, 1.0, and 1.8 standard deviations above the mean.
- Excessive growth is particularly pronounced during the first four years of life.
- Bone age is advanced and gradually increases until the fifth year, after which the difference between bone age and chronological age stabilizes.
- From four years onward, growth curves usually remain above the 97th centile.
- Midparental height of affected individuals is similar to the normal mean midparental height.
- A small percentage of affected individuals have more extreme final height attainment.
- Early feeding problems are observed in 35%.
- The hands and feet are large.
- Disharmonic maturation and abnormal sequences in the appearance of carpal bones occur in some affected individuals.
- A characteristic metacarpophalangeal pattern has been reported.
- Joint laxity is frequent with pes planus especially common.
- Rarely, there is significant scoliosis.
- Performance and CNS Abnormalities
Most patients have nonprogressive neurologic dysfunction manifested by unusual clumsiveness which improves with age.
Cole and Hughes reported a mean DQ/IQ of 78 with a range of 40–129 (n = 23), but indicated that their figures probably underestimated ability in Sotos syndrome because some children from regular schools could not be formally assessed.
Delay in expressive language and motor development during infancy is particularly common and in some instances may be followed by attainment of normal or near normal intelligence Opens in new window.
Cole and Hughes have observed that patients with Sotos syndrome tend to improve as they get older.
- Delay in walking until after 15 months of age and speech delay until after two and one half years are usual.
- Autism has been seen.
- Seizures Opens in new window and respiratory and feeding problems have been noted in about 20%.
- Often drooling is observed.
- Attention deficit may also be a component in some instances.
Occasionally, other neurological signs have been reported, including nystagmus, strabismus, increased deep tendon reflexes, hypotonia, and muscle weakness. Dilatation of the cerebral ventricles is seen in 70%–100%.
Other abnormalities include absent corpus callosum, prominent cortical sulci, cavum septum pellucidum, and cavum velum interpositi.
Schaefer et al in a neuroimaging study of 40 patients, found prominence of the trigone in 90%, prominence of the occipital horns in 75%, and ventriculomegaly in 63%.
- Craniofacial Features
Dolichocephaly and marked frontal bossing are accentuated by frontoparietal balding.
- The head circumference is usually well above the 97th centile.
- Narrow temples make the eyes appear wide-set, but true ocular hypertelorism is not found.
- Strabismus is noted in 40%.
- The cheeks are full.
Although the mandible is long and narrow inferiorly, squared, or pointed, true prognathism is rare. The palate is highly arched, and prematurely erupted deciduous teeth are observed in more than 50% of cases. Cephalometric analyses are available.
- Neoplasms
Cohen critically reviewed neoplasms reported to occur with Sotos syndrome. Retrospective literature review suggests a tumor frequency of about 3.9%.
- Tumors have included Wilms tumor,
- hepatocellular carcinoma,
- neuroblastoma,
- vaginal epidermoid carcinoma,
- small cell carcinoma of lung,
- sacrococcygeal teratoma,
- giant cell granuloma of the mandible, and
- acute lymphatic leukemia.
- Laboratory Findings
A 14% frequency of glucose intolerance has been demonstrated in Sotos syndrome by numerous investigators.
- Other Findings
Congenital heart defects have been discussed by Keneko et al, Cole and Hughes, Noreau et al, and Tsukahara et al. Many low-frequency abnormalities have been reported, including, among others,
- juvenile macular degeneration,
- optic disc pallor,
- glaucoma,
- bones in the anterior fontanelle,
- vertebra plana,
- kyphoscoliosis,
- brittle nails,
- syndactyly,
- functional megacolon, and
- autonomic failure with persistent fever.
Diagnosis and Differential Diagnosis
Most patients can be diagnosed by craniofacial gestalt accompanied by significant overgrowth.
However, enlarged head circumference may be seen in several other conditions including hydrocephalus, neurofibromatosis, achondroplasia, autosomal dominant macrocephaly, and so on.
Occasional patients have been confused as having Weaver syndrome Opens in new window. Still other patients cannot be confidently diagnosed as having either syndrome.
Opitz et al suggested that the many similarities between Sotos syndrome and Weaver syndrome may possibly indicate allelic rather than locus heterogeneity.
Boys with fragile X may overlap phenotypically with those having Sotos syndrome.
Although some syndromes occur with overgrowth and others are associated with macrocephaly Opens in new window, Sotos syndrome is a distinctive condition.
Several syndromes have been confused with Sotos syndrome. Cole and Hughes evaluating 79 patients, found that 50% were misdiagnosed.
Cole and Hughes and others proposed that autosomal dominant macrocephaly, obesity, unusual facies, delayed bone age, mental retardation and autism is a distinct entity. This has come to known as Cole-Hughes macrocephaly syndrome.
See also:
- Allanson JE, Cole TRP: Sotos syndrome: Evolution of facial phenotype. Subjective and objective assessment. Am J Med Genet 65:13-20, 1996.
- Beemer FA et al: Cerebral gigantism (Sotos syndrome) in two patients with fragile (X) syndrome. Am J Med Genet 23:221-226, 1986.
- Brown WT et al: Identical twins discordant for Sotos syndrome. Am J Med Genet 79:329-33, 1998.
- Butler MG et al: Metacarpophalangeal pattern profile analysis in Sotos syndrome. Am J Med Genet 20:625-629, 1985.
- Butler MG et al: Metacarpophalangeal profile analysis in Sotos syndrome: A follow-up report on 34 subjects. Am J Med Genet 29:143-147, 1988.
- Char F: Sotos syndrome from childhood to adolescence. J Clin Dysmorphol 2:29-52, 1984.
- Cohen MM Jr: A comprehensive and critical assessment of overgrowth and overgrowth syndromes. In: Advances in Human Genetics, Harris H and Hirschhorn K (eds), Plenum Press, New York, Vol 18, Ch 4, pp 181-303; Addendum, pp 373-376, 1989.
- Cohen MM Jr: Tumors and non-tumors in Sotos syndrome. Am J Med Genet 84:173-175, 1999.
- Cole TRP, Hughes HE: Sotos syndrome. J Med Genet 27:571-576, 1990.
- Cole TRP, Hughes HE: Sotos syndrome: A study of the diagnostic criteria and natural history. J Med Genet 31:20-32, 1994.
- Cole TRP et al: Small long cell carcinoma in a patient with Sotos syndrome: Are genees at 3p21 involved in both conditions? J Med Genet 29:338-341, 1992.
- Dijstra PF et al: Metacarpophalangeal pattern profile analysis in Sotos and Marfan syndrome. Am J Med Genet 51:55-60, 1994.
- Evans PR: Sotos’ syndrome (cerebral gigantism) with peripheral dysostosis. Arch Dis Child 46:199-202, 1971.
- Goldstein DJ et al: Overgrowth, congenital hypotonia, nystagmus, strabismus, and mental retardation: Variant of dominantly inherited Sotos sequence? Am J Med Genet 29:783-792, 1988.
- Haga N et al: Scoliosis in cerebral gigantism, Sotos syndrome. Spine 21:1699-1702, 1996.
- Hersh JH et al: Risk of malignancy in Sotos syndrome. J Pediatr 120:572-574, 1992.
- Inoue K et al: Optic disc pallor and retinal atrophy in Sotos syndrome. Am J Ophthalmol 130:853-854, 2000.
- Kok K et al: Breakpoint mapping by FISH in a Sotos patient with a constitutional translocation t(3;6). J Med Genet 36:346-347, 1999.
- Morrow JD et al: Autistic disorder in Sotos syndrome. Eur J Pediatr 149:567-569, 1990.
- Naqvi S et al: Cole-Hughes macrocephaly syndrome and associated autistic manifestations. Am J Med Genet 94:149-152, 2000.
