
Osteogenesis Imperfecta
Clinical Definition and Features
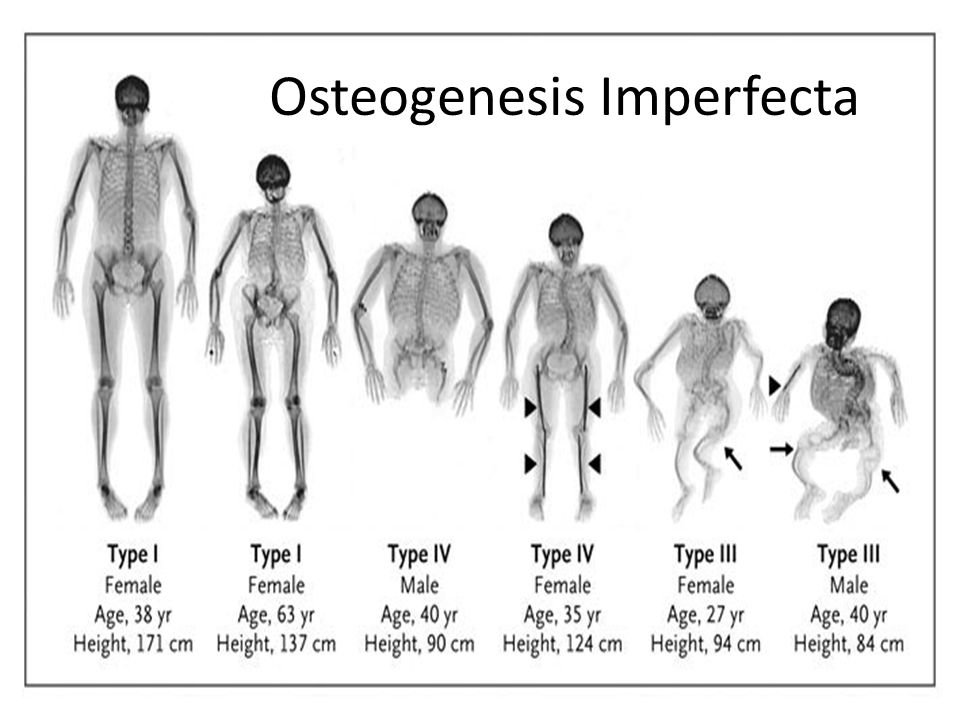 Figure X-1 | Image courtesy of OI Foundation Opens in new window
Figure X-1 | Image courtesy of OI Foundation Opens in new window
Osteogenesis imperfecta (OI) is a heterogeneous group of genetic disorders of type I collagen metabolism characterized by extreme fragility and porosity of the bones, with an attendant proneness to fracture.
The severity of OI symptoms ranges from prenatal death to mild osteopenia without limb deformity; and the incidence ranges from 1 per 20,000 to 1 per 50,000 live births. There is no predilection for race, gender, or ethnic origin.
Although bone fragility is the cardinal feature of osteogenesis imperfecta, other tissues in which type I collagen is the primary structural protein are also affected. These include skin, tendon, ligament, and dentin.
Associated features in some affected individuals include:
- blue sclera,
- opalescent teeth with characteristic radiologic features,
- hearing loss,
- deformity of long bones and spine, and
- joint hyperextensibility.
Types of Osteogenesis Imperfecta
Osteogenesis imperfecta is divided on a clinical and hereditary basis into four types. The severity of the disorder varies widely between and even within families; some individuals have minimum involvement of the skeleton and may never have a fracture, whereas others have very severe involvement and many fractures.
Clinical and genetic studies delineate four major syndrome groups (Table X-1). The estimated prevalence of all types combined is about 0.5/10,000 births (8).
Although all of these syndromes are likely heterogeneous (7) at the clinical, radiographic, and molecular levels.
| Table X-1 | Major osteogenesis imperfecta syndromes | |||
|---|---|---|---|
| Type | Salient features | Inheritance | Most common molecular background |
| Type I |
| AD (common) | Nonfunctional COL1A1 gene (null allele) |
| Type IA |
| ||
| Type IB |
| ||
| Type II |
|
| Point mutations resulting in substitution of Gly residues in triple helix domain of α1(I)/α2(I) |
| Table X-1 Continues | Major osteogenesis imperfecta syndromes | |||
|---|---|---|---|
| Type | Salient features | Inheritance | Most common molecular background |
| Type III |
|
| Deletions in COL1A1 gene causing frameshift |
| Type IV |
| AD | Point mutations in COL1A2 gene |
| Type IVA |
| ||
| Type IVB |
| ||
Nomenclature classifying patients into congenital and tarda forms is not useful, as individuals with any osteogenesis imperfect syndrome can be born with fractures; in addition, this feature cannot be consistently correlated with inheritance pattern, prognosis, or recurrence risk (1,2,4).
Classification into broad-boned and thin-boned types is also unsatisfactory, as change from one shape to another can occur with age.
Molecular pathology
The different forms of osteogenesis imperfecta are caused by mutations Opens in new window in the two genes that encode the chains of type I collagen—i.e., COL1A1, located on chromosome 17, and COL1A2, located on chromosome 7.
These proteins encode encode for the pro-α1 and pro-α2 chains of type 1 procollagen. Two pro-α1 chains and one pro-α2 chain together form one heterotrimer.
This procollagen is secreted by fibroblasts, after which the aminopropeptides on each of the three pro-α chains are cleaved off by a protease, and the carboxy-propeptides are cleaved off by another protease.
Thereafter, the collagen molecule self-assembles spontaneously to form fibril structures. The formation of fibrils depends heavily on the uniformity of the collagen molecules, and abnormal molecules have a deleterious effect on fibril structures.
Each of the collagen genes contain more than 50 exons that contain the coding sequence for proteins of about 1400 amino acids.
The primary sequence consists of a repeating tripeptide unit which can be written (Gly-X-Y). The amino acid glycine is in every third position, allowing for the tight helical structure because it is small and contains no side chains.
Hydroxyproline Opens in new window is found only in the Y position, which it occupies in about a third of the triplets, often preceded by proline. The first exons encode the signal sequence, followed by the protease cleavage site.
The final exons encode an additional protease cleavage site and the globular carboxyl-terminal domain. The stability of the triple helix is provided by interchain hydrogen bonds.
Clinical Manifestations
Variations in the clinical manifestations of osteogenesis imperfecta relate directly to the heterogeneity of genetic defects in type I collagen genes.
The number of fractures varies according to the severity of the disease. In general, the earlier in life the fractures occur, the more severe the disease.
Musculoskeletal abnormalities include long bone deformities with anterior bowing of the humerus, tibia, and fibula, and lateral bowing of the femur, radius, and ulna.
The femur bone Opens in new window is the most commonly fractured long bone, with the fracture usually located at the convexity of the bone appearing transverse and minimally displaced. It is not uncommon for children to complain of minimal pain, since there is usually minimal soft tissue injury and they are accustomed to frequent fractures.
Multiple fractures within the same bone often occur as a result of the severe angulation in which it heals and because of disuse atrophy, both of which make the bone more susceptible to a second fracture.
Bowing of the long bones results from multiple transverse fractures and the pulling of strong muscles. Cranial deformity is also common. There is flattening of the posterior cranium with a bulging calvaria and a triangular shaped face.
The forehead is usually broad with prominent parietal and temporal bones. Spinal deformities include such severe kyphoscoliosis that pulmonary complications are often seen.
The incidence of spiral deformities ranges from 90% for type II osteogenesis imperfecta Opens in new window to 10% to 40% for type I Opens in new window osteogenesis imperfecta. The most common spinal deformity is a thoracic scoliosis.
Ligamentous laxity results in hypermobility of the joints and frequent dislocations. Cubitus varus with flexion contractures at the elbow is another common finding.
Dislocations of the radial head, the hip joint, and the patellofemoral joint are also common occurrences increasing the incidence of falls and further fractures. Muscular hypotonia is seen secondary to ligamentous abnormalities and reduced activity.
Due to the deficiency of dentin, the teeth of some osteogenesis imperfecta patients are extremely brittle, breaking easily and becoming susceptible to caries.
The enamel is usually normal since it is of ectodermal, and not mesenchymal, origin. If the teeth are affected in osteogenesis imperfecta patients, this is referred to as dentinogenesis imperfecta Opens in new window, a condition that has been used by Sillence (1,2) to subclassify type I and type III osteogenesis imperfecta.
Other findings
Extraskeletal findings include blue sclera due to abnormal corioid, hearing loss, and growth retardation.
However, blue sclera, another hallmark characteristic of osteogenesis imperfect, is not found in all types. In type I Opens in new window, sclera are distinctly blue throughout life; and in type IV Opens in new window, the sclera are normal.
Deafness occurs in approximately 40% of type I osteogenesis imperfecta Opens in new window patients, with lower percentages in type IV Opens in new window.
Hearing loss usually begins at adolescence and worsens with age, and results from conductive loss due to otosclerosis of the ossicles or from neurosensory loss due to compression of the auditory nerve as it exits the skull. In addition to loss of hearing, patients may also complain of tinnitus and vertigo.
Radiographic findings include marked, generalized osteoporosis Opens in new window with thin cortices. Multiple stages of fracture healing reflect the numerous fractures sustained by these patients.
Malunions of these multiple fractures result in shortened long bones with severe bowing. The cortices are characteristically thin with occasional thickened areas secondary to callus formation. Fairbank (1948) described three types of radiographic findings:
- thick bone is seen at areas of prior fractures where there is large callus formation;
- thin bone appears with very narrow shafts, thin trabeculae, thin cortices, and marked osteopenia; and
- cystic bone is a complication of immobilization from fracture treatment.
The lack of ambulation and normal stress placed upon the bones to stimulate bone formation results in a cystic honeycomb pattern. Flaring of the metaphyses is present, indicating abnormal bone modeling.
See also:
- Therapeutics for Osteogenesis ImperfectaOpens in new window
- Clinical Features of Type I Osteogenesis ImperfectaOpens in new window
- Clinical Features of Type II Osteogenesis ImperfectaOpens in new window
- Clinical Features of Type III Osteogenesis ImperfectaOpens in new window
- Clinical Features of Type IV Osteogenesis ImperfectaOpens in new window
- Sillence DO et al: Osteogenesis imperfecta. An expanding panorama of variants. Clin Orthoped Rel Res 159:11-25, 1981.
- Sillence DO et al: Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 16:101-116, 1979.
- Silence DO et al: Clinical variability in osteogenesis imperfecta—variable expressivity or genetic heterogeneity. Birth Defects 15(5B): 113-129, 1979.
- Thompson EM et al: Recurrence risks and prognosis in severe sporadic osteogenesis imperfecta. J Med Genet 24:390-405, 1987.
- Bauze RJ et al: A new look at osteogenesis imperfect: A clinical, radiological and biochemical study of forty-two patients. J Bone Joint Surg Br 57:2-12, 1975.
- Spranger J: Osteogenesis imperfect: A pasture for splitters and lumpers. Am J Med Genet 17:425-428, 1984.
- Stoss H et al: Heterogeneity of osteogenesis imperfect. Biochemical and morphological findings in a case of type III according to Sillence. Eur J Pediatr 145:34-39, 1986.
- Stoll C et al: Birth prevalence rates of skeletal dysplasias. Clin Genet 35:88-92, 1989.
- Smith R et al: The eye and collagen in osteogenesis imperfecta. Birth Defects 12(3):563-566, 1976.
- Sillence DO et al: Natural history of blue sclerae in osteogenesis imperfecta. Am J Med Genet 45:183-186, 1993.
- Smith R et al: The Brittle Bone Syndrome. Osteogenesis Imperfecta. Butterworths, London, 1983.
- Carey MC et al: Osteogenesis imperfecta in twenty-three members of a kindred with heritable features contributed by a nonspecific skeletal disorder. Q J Med 37:437-449, 1968.
- Madigan WP et al: Retinal detachment in osteogenesis imperfecta. J Pediatr Ophthalmol Strabismus 31:268-269, 1994.
- Carpenter BF, Hunter AGW: Micromelia, polysyndactyly, multiple malformations, and fragile bones in a stillborn child. J Med Genet 19:311, 1982.
- Reidner ED et al: Hearing patterns in dominant osteogenesis imperfecta. Arch Otolaryngol 106:737-740, 1980.
- Cox JR, Simmons CL: Osteogenesis imperfecta and associated hearing loss in five kindreds. South Med J 75:1222-1226, 1982.
- Garretsen TJTM, Cremers CWRJ: Clinical and genetic aspects in autosomal dominant inherited osteogenesis imperfect type 1. Ann N Y Acad Sci 630:240-248, 1991.
- Hortop J et al: Cardiovascular involvement in osteogenesis imperfecta. Circulation 73:54-61. 1986.
- Pyeritz RE, Levin LS: Aortic root dialatation and valvular dysfunction in osteogenesis imperfecta. Circulation 64:1193A, 1981.
- White NJ et al: Cardiovascular abnormalities in osteogenesis imperfect. Am Heart J 106:1416-1420, 1983.
- Paterson CR et al: Osteogenesis imperfecta with dominant inheritance and normal sclerae. J Bone Joint Surg Br 65:35-39, 1983.
- Paterson CR et al: Osteogenesis imperfect after the menopause. N Engl J Med 310:1694-1696, 1984.
- Gillerot Y et al: Lethal perinatal type II osteogenesis imperfect in a family with a dominantly inherited type I. Eur J Pediatr 141:119-122, 1983.
- Heyes FM et al: Osteogenesis imperfecta and odontogenesis imperfecta: Clinical and genetic aspects in eighteen families. J Pediatr 56:234-245, 1960.
- Levin LS et al: The dentition in the osteogenesis imperfecta syndromes. Clin Orthop Rel Res 159:64-74, 1981.
- Lukinmaa P-L et al: Dental findings in osteogenesis imperfecta I. occurrence and expression of type I dentinogenesis imperfecta. J Craniofac Genet Dev Biol 7:115-125, 1987.
