
Incontinentia Pigmenti
Clinical Definition and Overview
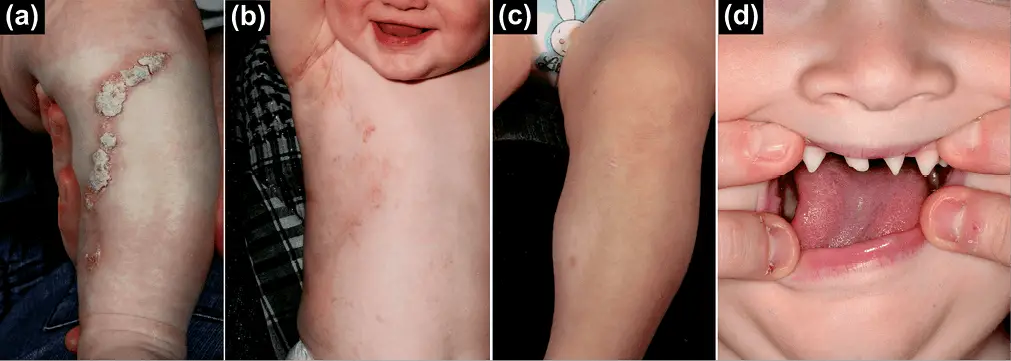
|
Incontinentia pigmenti (IP), formerly called incontinentia pigmenti type II or Bloch-Sulzberger syndrome, is a rare X-linked dominant disorder associated with a variety of abnormalities of the skin, hair, teeth, nails and central nervous system, which is prevalent primarily in females and occasionally seen in boys. IP is a multisystem disorder that may present to neonatologists, neurologists, ophthalmologists or dentists as well as dermatologists. However, the diagnosis focuses on the cutaneous findings. The name refers to the pathologic finding of pigmentary incontinence (i.e., dermal melanophages) in the third stage of the disease. |
Affected females typically have an erythematous, vesicular rash that appears at birth or soon after birth. This rash evolves over time, becoming verrucous and pigmented, and then atrophic. Adults have areas of linear hypopigmentation.
Other manifestations include alopecia, hypodontia/misshapen teeth, leukocytosis with eosinophilia, vascular abnormalities of the retina, and other eye findings.
Skeletal and dental anomalies are common and quite varied. Neurologic manifestations are primarily seizures and mental retardation.
Clinical recognition of IP began during the first decade of the 20th century. Garrod briefly described a young girl with severe neurodevelopmental deficits who had peculiar skin pigmentation consisting of linear streaks and whorls of hyperpigmentation (Garrod, 1906).
A more detailed description of incontinentia pigmenti was provided by Sulzberger in 1928, who noted the association of the skin pattern with other organ involvement in a patient reported 2 years earlier by Bloch, who introduced the term incontinentia pigmenti.
2. Clinical Manifestations
2.1 Skin
The skin lesions are the hallmark of IP. Affected females display skin abnormalities in four stages (below) evolve over weeks to years. The onset and duration of stage vary among patients; not all patients experience all four stages.
Skin lesions may recur or they may wax and wane in severity, and lesions of more than one stage can be present at a given time. This in combination with other factors make the diagnosis of IP difficult in atypical cases.
The skin abnormalities that define each stage occur along lines of embryonic and fetal skin development known as Blaschko’s lines—which corresponds with cell migration embrogenesis. Like dermatomes, they are linear on the limbs and circumferential on the trunk.
- Stage 1: The Bullous Stage
This is characterized by blister-like bullous eruptions that are linear on the extremities and/or circumferential on the trunk. The eruption can be erythematous and may appear infectious, though they are not. Stage 1 manifests within the first 6–8 weeks of life and can be present at birth. The bullous rash may relapse or worsen with fevers as late as one year of age. Rarely the blisters may not appear until after the first year of life.
- Stage 2: The Verrucous Stage
This is characterized by a hypertrophic, wart-like rash that is linear on the extremities and/or circumferential on the trunk. This stage usually manifests within the first few months of life. Occasionally it can be present at birth but more typically arises as stage 1 begins to resolve. Usually stage 2 persists for a few months, but it can persist for years. Stage 2 may also include the appearance of dystrophic nails and abnormalities of tooth eruption.
- Stage 3: The Hyperpigmentation Stage
This stage is characterized by macular, slate gray or brown hyperpigmentaton that occurs in a ‘marble cake’ or swirled pattern along Blaschko’s lines, usually circumferential on the trunk and linear on the extremities.
The hyperpigmentation stage is the most characteristic stage for IP. Not all females have extensive hyperpigmentation; it can in fact be quite limited. The most frequently involved areas are the groin and axilla.
The entire skin surface may need to be examined to find characteristic patterns. Hyperpigmentation begins at anywhere from 6 months to 1 year of age, usually as stage 2 begins to resolve. It is not usually present at birth and it persists into adulthood.
Usually, the hyperpigmentation begins to fade in the second or third decade of life. Patients older than teenagers may show no skin changes associated with IP. This is important to keep in mind when examining mothers of children with suspected IP.
- Stage 4: The Atretic Stage
This stage is characterized by linear hypopigmentation and alopecia, particularly noticeable on the extremities. The definition of this state remains open.
There may not be true hypopigmentation, but rather a loss of hair and epidermal glands. As with the three stages, the pattern follows Blaschko’s lines. This stages almost always does not occur in all patients. When present, it arises after the hyperpigmentation fades.
2.2 Hair
Alopecia occurs on the scalp, especially at the vertex, and the trunk and extremities. Patchy alopecia of the scalp may corresponds to areas of scarring left from blistering in stage 1, but may also occur in patients who have had no stage 1 or 2 lesions on the scalp.
Alopecia Opens in new window occurs in areas of skin hypopigmentation as part of stage 4 skin changes. Scalp hair may be thin in early childhood. Hair may also be lusterless, wiry, and coarse, often at the vertex in a ‘woolly-hair nevus. Eyebrows may be absent or sparse.
2.3 Breast
Abnormalities of mammary tissue ranging from aplasia of the breast to supernumerary nipples may be present. No consistent pattern has been observe.
2.4 Teeth
Dentition is affected in about two-thirds of patients with IP. Abnormalities include hypodontia, microdontia, abnormally-shaped teeth such as peg-shaped teeth, delayed eruption, malpositioning, and impaction. Enamel and tooth strength are normal. Orthodontic intervention may be required, and dental evaluation should be part of the overall management of a child with IP.
2.5 Nails
Nails can be streaked, pitted, or brittle. These dystrophic changes are usually seen during stages 2 and at times can be confused with fungal infections of the nails. They are often transient but may relapse.
2.6 Skeletal
Infrequently seen are a wide variety of skeletal disorders such as extra ribs, hemivertebrae, skull deformities, and chondrodystrophy. Scoliosis is a long-term potential complication of IP. Limb anomalies including club foot and syndactyly are uncommon. Hemiatrophy has been reported rarely.
2.7 Ophthalmologic
About a third of patients with IP have retinal or ocular pathology. Ocular anomalies include cataracts, strabismus, ptosis, and conjuctival hyperpigmentation. Bilateral retinoblastoma has been reported. A diffuse mottled hypopigmentation of the retina is said to be pathognomonic of IP.
The most significant medical problem associated with IP is the increased risk of retinal detachment, which is most likely to occur in infancy and childhood. Retinal detachment almost never occurs after age six years.
Retinal detachment is preceded by neovascularization in the peripheral retina, which is often followed by exudation and/or fibrosis, and thus children with arteriovenous anastomoses and associated preretinal fibrous changes are at high risk for this.
These changes are visible on examination through a dilated pupil using indirect ophthalmoscopy. The retinal neovascularization is similar to that seen in retinopathy.
If the diagnosis is suspected in the newborn nursery, a pediatric ophthalmologist should examine the retina to look for the typical pathologic findings before the baby leaves the hospital.
2.8 Peripheral Blood
Peripheral leukocytosis with up to 65% eosinophils may occur, particularly in stages 1 and 2. The eosinophil count typically peaks during the first few months of life. The cause of the leukocytosi is unknown, but it may be a reaction to tissue necrosis rather than a primary manifestation of the disease.
2.9 Neurological
The majority of patients with IP, both male and female, are intellectually normal.
Still, the incidence of mental retardation Opens in new window and developmental delay is higher in males than females who meet the IP diagnostic criteria.
Similar estimates are not available for females with IP; however, the risk seems to be low. In one study of 30 multigenerational families with over 100 affected females, only one affected child was known to be severely mentally retarded.
In another study only 3 of 15 children with IP were developmentally delayed, and one of these children had Down syndrome Opens in new window. A similar incidence of mental retardation was found in an earlier series.
Whether a correlation exists between any of the other signs of IP and central nervous system (CNS) involvement is unclear, but optic atrophy and severe retinal vaso-occlusive disease may be a marker for more severe neurodevelopmental deficits.
3. Genetics
Incontinentia pigmenti (IP) is an X-linked dominant disorder Opens in new window, usually antenatally lethal in boys, that is caused by mutations in the NEMO gene at Xq28. About half of patients with IP have clinically affected family members. Male fetuses with mutations of NEMO usually die in utero.
Less deleterious mutations can allow survival of males, but with ectodermal dysplasia and immunodeficiency. Male patients with skin, dental, and ocular abnormalities typical of those seen in female patients with IP (without immunodeficiency) are rare. Clinically affected males have either an extra wild type copy of the NEMO gene (Klinefelter’s 46XXY) or are somatic mosaics.
4. Pathogenetics
The NEMO protein is a subunit of a kinase that activates NF-kB, a transcription factor that protects against TNF-α-inducced apoptosis. The idea of IP as a pro-apoptotic state explains male lethality, destruction of epidermal cells, and progressive replacement of cells expressing the mutant X chromosome by those expressing the normal allele Opens in new window.
In one report, a liveborn boy whose mother had classic IP died hours after birth due to lethal hematopoietic and immunologic disturbances. Milder NEMO mutations are responsible for hypohidrotic ectodermal dysplasia with immune deficiency (HED-ID) in boys, whose mothers often have relatively subtle features of IP.
5. Therapeutics
Baseline and longitudinal ophthalmologic (especially important during infancy) and neurologic evaluations are recommended, as are dental assessments and early intervention when anomalies are present.
Treatment of the skin in an affected newborn is aimed at reducing the risk of infection of the bullous lesions. When infection occurs, it should be treated like any other cellulitis.
Topical medications, oatmeal baths, etc., may be useful to relieve discomfort. No treatments are known to shorten or lessen the cutaneous manifestations of stages 1 and 2 of the disease.
The hyperpigmentation of stage 3 fades with time and can be covered with cosmetics and clothing if necessary. Scalp alopecia can be of cosmetic concern. The parents of affected children should have a careful skin examination for atrophic, hypopigmented streaks and a retinal examination. Siblings should also have a skin examination.
It is recommended that children be referred to an ophthalmologist familiar with IP and diseases of the retina as soon as the diagnosis of IP is considered or established.
The parents should be instructed about the possibility of retinal detachment; any apparent changes in vision or any evidence of acquired strabismus should be evaluated promptly.
Routine eye examinations for children with IP are recommended every six to twelve months, depending on the severity of eye disease. The risk for retinal detachment decreases with age, and after about age six years annual eye examinations are recommended as part of preventative care. Laser photocoagulation is often effective in controlling the neovascularization.
Severe retinal disease is often associated with brain dysfunction and is a marker to pursue X-ray scanning studies of the head. With respect to the eyes themselves, some babies with IP and even some older patients might benefit from laser treatment or freezing therapy (cryopexy) in an effort to prevent retinal detachment or vitreous hemorrhage from the consequences of the typical retinal neovascularization that occurs in this disorder.
Ongoing dental evaluation should be obtaine. Most patients may consider cosmetic dentistry. Delayed or inadequate eruption of primary teeth can interfere with development of speech and with nutrition because of difficulty with chewing.
See also:
- Bloch, B. (1926). Eigentumliche, bisher nicht beschriebene Pigmentaffektion (incontinentia pigmenti). Schweizerische Medizinische Wochenschrift, 7:404-405.
- Brunquell, P. J. (1987). Recurrent encephalomyelitis associated with incontinentia pigmenti. Pediatric Neurology, 3: 174-177.
- Carney, R. G. (1976). Incontinentia pigmenti. A world statistical analysis. Archives of Dermatology, 112:535-542.
- Garrod, A. E. (1906). Peculiar skin pigmentation of the skin in an infant. Transactions of the Clinical Society of London, 39:216-217.
- Goldberg, M. F. & Custis, P. H. (1993). Retinal and other manifestations of incontinentia pigmenti (Bloch-Sulzberger syndrome). Ophtalmology, 100: 1645-1654.
- Heathcote, J. G., Schoales, B. A. & Willis, N. R. (1991). Incontinentia pigmenti (Bloch-Sulzberger syndrome): a case report and review of the ocular pathological features. Canadian Journal of Ophthalmology, 26:229-237.
- Kasmann-Kellner, B., Jurin-Bunte, B. & Ruprecht, K. W. (1999). Incontinentia pigmenti (Bloch-Sulzberger-syndrome): case report and differential diagnosis to related dermato-ocular syndromes. Ophthalmologica, 213:63-69.
- O’Brien, J. E. & Feingold, M. (1985). Incontinentia pigmenti. A longitudinal study. American Journal of Diseases Childhood, 139: 711-712.
- Siemes, H., Schneider, H., Dening, D. & Hanefeld, F. (1978). Encephalitis in two members of a family with incontinentia pigmenti (Bloch-Sulzberger syndrome). The possible role of inflammation in the pathogenesis of CNS involvement. European Journal of Pediatrics, 129: 103-115.
- Sulzberger, M. B. (1928). Uber eine bisher nicht beschriebene congenital Pigmentanomalie (incontinentia pigmenti). Archives of Dermatology and Syphilis (Berlin), 154:19-32.
